I am delighted to share that me, Joel A. Tropp, and Robert J. Webber‘s paper XTrace: Making the Most of Every Sample in Stochastic Trace Estimation has recently been released as a preprint on arXiv. In it, we consider the implicit trace estimation problem:
Implicit trace estimation problem: Given access to a square matrix  via the matrix–vector product operation
via the matrix–vector product operation  , estimate its trace
, estimate its trace  .
.
Algorithms for this task have many uses such as log-determinant computations in machine learning, partition function calculations in statistical physics, and generalized cross validation for smoothing splines. I described another application to counting triangles in a large network in a previous blog post.
Our paper presents new trace estimators XTrace and XNysTrace which are highly efficient, producing accurate trace approximations using a small budget of matrix–vector products. In addition, these algorithms are fast to run and are supported by theoretical results which explain their excellent performance. I really hope that you will check out the paper to learn more about these estimators!
For the rest of this post, I’m going to talk about the most basic stochastic trace estimation algorithm, the Girard–Hutchinson estimator. This seemingly simple algorithm exhibits a number of nuances and forms the backbone for more sophisticated trace estimates such as Hutch++, Nyström++, XTrace, and XNysTrace. Toward the end, this blog post will be fairly mathematical, but I hope that the beginning will be fairly accessible to all.
Girard–Hutchinson Estimator: The Basics
The Girard–Hutchinson estimator for the trace of a square matrix is
(1) ![\[\hat{\tr} = \frac{1}{m} \sum_{i=1}^m \omega_i^* A \omega_i. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a287886ee24c9ab99dfe35b6cdae83cf_l3.png "Rendered by QuickLaTeX.com")
Here,  are random vectors, usually chosen to be statistically independent, and
are random vectors, usually chosen to be statistically independent, and  denotes the conjugate transpose of a vector or matrix. The Girard–Hutchinson estimator only depends on the matrix through the matrix–vector products
denotes the conjugate transpose of a vector or matrix. The Girard–Hutchinson estimator only depends on the matrix through the matrix–vector products  .
.
Unbiasedness
Provided the random vectors are isotropic
(2) ![\[\mathbb{E} [\omega_i\omega_i^*] = I, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-74bb76367ad1408bd538d049774cf990_l3.png "Rendered by QuickLaTeX.com")
the Girard–Hutchinson estimator is unbiased:
(3) ![\[\mathbb{E} [\hat{\tr}] = \tr A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f6407c7a741003cc20780e72d050cb56_l3.png "Rendered by QuickLaTeX.com")
Let us confirm this claim in some detail. First, we use linearity of expectation to evaluate
(4) ![\[\mathbb{E} [\hat{\tr}] = \mathbb{E} \left[ \frac{1}{m} \sum_{i=1}^m \omega_i^*A\omega_i \right] = \frac{1}{m} \sum_{i=1}^m \mathbb{E} \left[ \omega_i^* A \omega_i\right]. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-52eebd003a597cac5b2c69f7b6e537e8_l3.png "Rendered by QuickLaTeX.com")
Therefore, to prove that ![\mathbb{E} [\hat{\tr}] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-96f71a1b054ffed9e316cf9cb7a16c5c_l3.png "Rendered by QuickLaTeX.com") , it is sufficient to prove that
, it is sufficient to prove that ![\mathbb{E} \left[\omega_i^*A\omega_i\right] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-120169dca277175c51f09b6f320cf5fa_l3.png "Rendered by QuickLaTeX.com") for each
for each  .
.
When working with traces, there are two tricks that solve 90% of derivations. The first trick is that, if we view a number as a  matrix, then a number equals its trace,
matrix, then a number equals its trace,  . The second trick is the cyclic property: For a
. The second trick is the cyclic property: For a  matrix
matrix  and a
and a  matrix
matrix  , we have
, we have  . The cyclic property should be handled with care when one works with a product of three or more matrices. For instance, we have
. The cyclic property should be handled with care when one works with a product of three or more matrices. For instance, we have
![\[\tr[BCD] = \tr[(BC)D] = \tr[D(BC)] = \tr[DBC].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cc1cd4920a2431e93b5453b5373999e3_l3.png "Rendered by QuickLaTeX.com")
However,
![\[\tr [BCD] \ne \tr[CBD] \quad \text{in general}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f563660fc58383a37ae7bedea16b2b9c_l3.png "Rendered by QuickLaTeX.com")
One should think of the matrix product  as beads on a closed loop of string. One can move the last bead
as beads on a closed loop of string. One can move the last bead  to the front of the other two,
to the front of the other two, ![\tr [BCD] = \tr[DBC]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7656627b133add21ab8765532def07d6_l3.png "Rendered by QuickLaTeX.com") , but not interchange two beads,
, but not interchange two beads, ![\tr[BCD] \ne \tr[CBD]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0f43e4f72510a69b596f7b9f7504dd58_l3.png "Rendered by QuickLaTeX.com") .
.
With this trick in hand, let’s return to proving that for every . Apply our two tricks:
![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[A\omega_i\omega_i^*\right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b1bb373766e64179bc0635f058a07f3f_l3.png "Rendered by QuickLaTeX.com")
The expectation is a linear operation and the matrix is non-random, so we can bring the expectation into the trace as
![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[A\omega_i\omega_i^*\right] = \tr(A \mathbb{E}[\omega_i\omega_i^*] ).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1d10f835c99efab005f0ad12cdb5647b_l3.png "Rendered by QuickLaTeX.com")
Invoke the isotropy condition (2) and conclude:
![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \tr(A \mathbb{E}[\omega_i\omega_i^*] ) = \tr(A\cdot I) = \tr A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b51781498146df3547c1503e451ad682_l3.png "Rendered by QuickLaTeX.com")
Plugging this into (4) confirms the unbiasedness claim (3).
Variance
Continue to assume that the  ‘s are isotropic (3) and now assume that are independent. By independence, the variance can be written as
‘s are isotropic (3) and now assume that are independent. By independence, the variance can be written as
![\[\Var(\hat{\tr}) = \frac{1}{m^2} \sum_{i=1}^m \Var(\omega_i^*A\omega_i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d9398dc43178ba9bb72aead0214e77c8_l3.png "Rendered by QuickLaTeX.com")
Assuming that are identically distributed  , we then get
, we then get
![\[\Var(\hat{\tr}) = \frac{1}{m} \Var(\omega^*A\omega).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4501a18842234362f8f99d523c7c723e_l3.png "Rendered by QuickLaTeX.com")
The variance decreases like  , which is characteristic of Monte Carlo-type algorithms. Since
, which is characteristic of Monte Carlo-type algorithms. Since  is unbiased (i.e, (3)), this means that the mean square error decays like so the average error (more precisely root-mean-square error) decays like
is unbiased (i.e, (3)), this means that the mean square error decays like so the average error (more precisely root-mean-square error) decays like
![\[\left| \hat{\tr} - \tr A \right| \lessapprox \frac{\mathrm{const}}{\sqrt{m}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c735791b8fe88d627bc2ebad8137fb1e_l3.png "Rendered by QuickLaTeX.com")
This type of convergence is very slow. If I want to decrease the error by a factor of  , I must do
, I must do  the work!
the work!
Variance-reduced trace estimators like Hutch++ and our new trace estimator XTrace improve the rate of convergence substantially. Even in the worst case, Hutch++ and XTrace reduce the variance at a rate  and (root-mean-square) error at rates :
and (root-mean-square) error at rates :
![\[\Var(\hat{\tr}_{\text{H++ or X}}) \le \frac{\mathrm{const}}{m^2},\quad \left| \hat{\tr}_{\text{H++ or X}} - \tr A \right| \lessapprox \frac{\mathrm{const}}{m}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-87438d87094d3df389400b5f7abee24a_l3.png "Rendered by QuickLaTeX.com")
For matrices with rapidly decreasing singular values, the variance and error can decrease much faster than this.
Variance Formulas
As the rate of convergence for the Girard–Hutchinson estimator is so slow, it is imperative to pick a distribution on test vectors  that makes the variance of the single–sample estimate
that makes the variance of the single–sample estimate  as low as possible. In this section, we will provide several explicit formulas for the variance of the Girard–Hutchinson estimator. Derivations of these formulas will appear at the end of this post. These variance formulas help illuminate the benefits and drawbacks of different test vector distributions.
as low as possible. In this section, we will provide several explicit formulas for the variance of the Girard–Hutchinson estimator. Derivations of these formulas will appear at the end of this post. These variance formulas help illuminate the benefits and drawbacks of different test vector distributions.
To express the formulas, we will need some notation. For a complex number  we use
we use  and
and  to denote the real and imaginary parts. The variance of a random complex number
to denote the real and imaginary parts. The variance of a random complex number  is
is
![\[\Var(z) := \mathbb{E} |z - \mathbb{E} z|^2 = \Var(\Re z) + \Var(\Im z).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8f636667a7dbbb3217896a962de1d28c_l3.png "Rendered by QuickLaTeX.com")
The Frobenius norm of a matrix is
![\[\left\|A\right\|_{\rm F}^2 = \sum_{i,j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e132e4b9029c41dbfb99cd0e588d8d70_l3.png "Rendered by QuickLaTeX.com")
If is real symmetric or complex Hermitian with (real) eigenvalues  , we have
, we have
(5) ![\[\left\|A\right\|_{\rm F}^2 = \sum_{i=1}^n \lambda_i^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06b272e602552353c020f6399347c748_l3.png "Rendered by QuickLaTeX.com")
 denotes the ordinary transpose of and
denotes the ordinary transpose of and  denotes the conjugate transpose of .
denotes the conjugate transpose of .
Real-Valued Test Vectors
We first focus on real-valued test vectors . Since is real, we can use the ordinary transpose  rather than the conjugate transpose . Since
rather than the conjugate transpose . Since  is a number, it is equal to its own transpose:
is a number, it is equal to its own transpose:
![\[\omega^\top A \omega = (\omega^\top A \omega)^\top = \omega^\top A^\top \omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3d543dc0d4d64d3c28f0f291cd9c8e83_l3.png "Rendered by QuickLaTeX.com")
Therefore,
![\[\omega^\top A\omega = \frac{\omega^\top A \omega + \omega^\top A^\top \omega}{2} = \omega^\top \left( \frac{A + A^\top}{2} \right)\omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f1bbe4f0c983f73e2461b593b752f54a_l3.png "Rendered by QuickLaTeX.com")
The Girard–Hutchinson trace estimator applied to is the same as the Girard–Hutchinson estimator applied to the symmetric part of ,  .
.
For the following results, assume is symmetric,  .
.
- Real Gaussian: are independent standard normal random vectors.
![\[\Var(\omega^\top A\omega) = 2 \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-091cf638fab943be5fd224acdb42668f_l3.png "Rendered by QuickLaTeX.com")
- Uniform signs (Rademachers): are independent random vectors with uniform
 coordinates.
coordinates. ![\[\Var(\omega^\top A \omega) = 2\sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-74621dad79cb3f06a8b70c661df4e01d_l3.png "Rendered by QuickLaTeX.com")
- Real sphere: Assume
 are uniformly distributed on the real sphere of radius
are uniformly distributed on the real sphere of radius  :
:  .
. ![\[\Var(\omega^\top A\omega) = \frac{2n}{n+2} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n} |\tr A|^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1e419b8da9b953a0b8b914df9e89f8c6_l3.png "Rendered by QuickLaTeX.com")
These formulas continue to hold for nonsymmetric by replacing by its symmetric part on the right-hand sides of these variance formulas.
Complex-Valued Test Vectors
We now move our focus to complex-valued test vectors . As a rule of thumb, one should typically expect that the variance for complex-valued test vectors applied to a real symmetric matrix is about half the natural real counterpart—e.g., for complex Gaussians, you get about half the variance than with real Gaussians.
A square complex matrix has a Cartesian decomposition
![\[A = A^{\rm H} + i A^{\rm SH}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d61da43cd6497ceefd2306aabb52ee67_l3.png "Rendered by QuickLaTeX.com")
where
![\[A^{\rm H} = \frac{A+A^*}{2} ,\quad A^{\rm SH} = \frac{A - A^*}{2i}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc287a49a59dca9421fdd2bef6be5334_l3.png "Rendered by QuickLaTeX.com")
denote the Hermitian and skew-Hermitian parts of . Similar to how the imaginary part of a complex number is real, the skew-Hermitian part of a complex matrix is Hermitian (and  is skew-Hermitian). Since
is skew-Hermitian). Since  and
and  are both Hermitian, we have
are both Hermitian, we have
![\[\Re(\omega^* A\omega) = \omega^* A^{\rm H} \omega, \quad \Im (\omega^* A \omega) = \omega^* A^{\rm SH} \omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6809a35cd06d92218ce8d597847f74bd_l3.png "Rendered by QuickLaTeX.com")
Consequently, the variance of  can be broken into Hermitian and skew-Hermitian parts:
can be broken into Hermitian and skew-Hermitian parts:
![\[\Var(\omega^* A\omega) = \Var(\omega^* A^{\rm H}\omega) + \Var(\omega^* A^{\rm SH}\omega).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e4658c38df479c5805dabd94b64cc0b3_l3.png "Rendered by QuickLaTeX.com")
For this reason, we will state the variance formulas only for Hermitian , with the formula for general following from the Cartesian decomposition.
For the following results, assume is Hermitian,  .
.
- Complex Gaussian: are independent standard complex random vectors, i.e., each has iid entries distributed as
 for
for  standard normal random variables.
standard normal random variables. ![\[\Var(\omega^* A\omega) = \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1beb66e032d83431dac6f8e0d6655eec_l3.png "Rendered by QuickLaTeX.com")
- Uniform phases (Steinhauses): are independent random vectors whose entries are uniform on the complex unit circle
 .
. ![\[\Var(\omega^* A \omega) = \sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1cb0e8df66c34056cd350752070af17a_l3.png "Rendered by QuickLaTeX.com")
- Complex sphere: Assume are uniformly distributed on the complex sphere of radius :
 .
. ![\[\Var(\omega^* A\omega) = \frac{n}{n+1} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n} |\tr A|^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-aacfa2afb45905f6abe0697c769cd1b7_l3.png "Rendered by QuickLaTeX.com")
Optimality Properties
Let us finally address the question of what the best choice of test vectors is for the Girard–Hutchinson estimator. We will state two results with different restrictions on .
Our first result, due to Hutchinson, is valid for real symmetric matrices with real test vectors.
Optimality (independent test vectors with independent coordinates). If the test vectors  are isotropic (2), independent from each other, and have independent entries, then for any fixed real symmetric matrix , the minimum variance for is obtained when are populated with random signs
are isotropic (2), independent from each other, and have independent entries, then for any fixed real symmetric matrix , the minimum variance for is obtained when are populated with random signs  .
.
The next optimality results will have real and complex versions. To present the results for  -valued and an
-valued and an  -valued test vectors on unified footing, let
-valued test vectors on unified footing, let  denote either or . We let a -Hermitian matrix be either a real symmetric matrix (if
denote either or . We let a -Hermitian matrix be either a real symmetric matrix (if  ) or a complex Hermitian matrix (if
) or a complex Hermitian matrix (if  ). Let a -unitary matrix be either a real orthogonal matrix (if ) or a complex unitary matrix (if ).
). Let a -unitary matrix be either a real orthogonal matrix (if ) or a complex unitary matrix (if ).
The condition that the vectors have independent entries is often too restrictive in practice. It rules out, for instance, the case of uniform vectors on the sphere. If we relax this condition, we get a different optimal distribution:
Optimality (independent test vectors). Consider any set  of -Hermitian matrices which is invariant under -unitary similary transformations:
of -Hermitian matrices which is invariant under -unitary similary transformations:
![\[\text{If $A \in \mathscr{A}$ and $U$ is $\field$-unitary, then $U^*AU \in \mathscr{A}$.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-18b4216d44b2364ea39ed4a078a067af_l3.png "Rendered by QuickLaTeX.com")
Assume that the test vectors are independent and isotropic (2). The worst-case variance  is minimized by choosing uniformly on the -sphere:
is minimized by choosing uniformly on the -sphere:  .
.
More simply, if you wants your stochastic trace estimator to be effective for a class of inputs (closed under -unitary similarity transformations) rather than a single input matrix , then the best distribution are test vectors drawn uniformly from the sphere. Examples of classes of matrices include:
- Fixed eigenvalues. For fixed real eigenvalues
 , the set of all -Hermitian matrices with these eigenvalues.
, the set of all -Hermitian matrices with these eigenvalues. - Density matrices. The class of all trace-one psd matrices.
- Frobenius norm ball. The class of all -Hermitian matrices of Frobenius norm at most 1.
Derivation of Formulas
In this section, we provide derivations of the variance formulas. I have chosen to focus on derivations which are shorter but use more advanced techniques rather than derivations which are longer but use fewer tricks.
Real Gaussians
First assume is real. Since is real symmetric, has an eigenvalue decomposition  , where
, where  is orthogonal and
is orthogonal and  is a diagonal matrix reporting ‘s eigenvalues. Since the real Gaussian distribution is invariant under orthogonal transformations,
is a diagonal matrix reporting ‘s eigenvalues. Since the real Gaussian distribution is invariant under orthogonal transformations,  has the same distribution as
has the same distribution as  . Therefore,
. Therefore,
![\[\Var(\omega^\top A \omega) = \Var(\omega^\top \Lambda \omega) = \Var \left( \sum_{i=1}^n \lambda_i \omega_i^2 \right) = \sum_{i=1}^n \lambda_i^2 \Var(\omega_i^2) = 2\sum_{i=1}^n \lambda_i^2 = 2\left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a7416823720bac922fae7cc685892a49_l3.png "Rendered by QuickLaTeX.com")
Here, we used that the variance of a squared standard normal random variable is two.
For non-real matrix, we can break the matrix into its entrywise real and imaginary parts  . Thus,
. Thus,
![\[\Var(\omega^\top A \omega) = \Var(\omega^\top \mathfrak{R}(A) \omega) + \Var(\omega^\top \mathfrak{I}(A) \omega) = 2\left\|\mathfrak{R}(A)\right\|_{\rm F}^2 + 2\left\|\mathfrak{I}(A)\right\|_{\rm F}^2 = 2\left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-527980eb3510f2c015106e6945126d41_l3.png "Rendered by QuickLaTeX.com")
Uniform Signs
First, compute
![\[\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega] = \sum_{i,j=1}^n A_{ij} \omega_i\omega_j - \sum_{i=1}^n A_{ii} = \sum_{i\ne j} A_{ij} \omega_i\omega_j + \sum_{i=1}^n A_{ii}(\omega_i^2-1).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4489cc9cb97ee7a0896bc61193d79910_l3.png "Rendered by QuickLaTeX.com")
For a vector of uniform random signs, we have  for every , so the second sum vanishes. Note that we have assumed symmetric, so the sum over
for every , so the second sum vanishes. Note that we have assumed symmetric, so the sum over  can be replaced by two times the sum over
can be replaced by two times the sum over  :
:
![\[\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega] = 2\sum_{i< j} A_{ij} \omega_i\omega_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-afc5f6bb40cd00b7897f25d73751f4ca_l3.png "Rendered by QuickLaTeX.com")
Note that  are pairwise independent. As a simple exercise, one can verify that the identity
are pairwise independent. As a simple exercise, one can verify that the identity
![\[\Var(a_1 X_1+\cdots+a_kX_k) = |a_1|^2 \Var(X_1) + \cdots + |a_k|^2 \Var(X_k)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b6fd81d15f564f514f79ab4c33d133d8_l3.png "Rendered by QuickLaTeX.com")
holds for any pairwise independent family of random variances  and numbers
and numbers  . Ergo,
. Ergo,
![\begin{align*}\Var(\omega^\top A\omega) &= \Var(\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega]) \\&= \Var\left(\sum_{i< j} 2A_{ij} \omega_i\omega_j\right) \\&= \sum_{i<j} 4 |A_{ij}|^2 \Var(\omega_i\omega_j) \\&= \sum_{i<j} 4 |A_{ij}|^2 \\&= 2 \sum_{i\ne j} |A_{ij}|^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bbb33f378fabd391c28d938420d3a32b_l3.png "Rendered by QuickLaTeX.com")
In the second-to-last line, we use the fact that  is a uniform random sign, which has variance
is a uniform random sign, which has variance  . The final line is a consequence of the symmetry of .
. The final line is a consequence of the symmetry of .
Uniform on the Real Sphere
The simplest proof is I know is by the “camel principle”. Here’s the story (a lightly edited quotation from MathOverflow):
A father left 17 camels to his three sons and, according to the will, the eldest son was to be given a half of the camels, the middle son one-third, and the youngest son the one-ninth. The sons did not know what to do since 17 is not evenly divisible into either two, three, or nine parts, but a wise man helped the sons: he added his own camel, the oldest son took  camels, the second son took
camels, the second son took  camels, the third son
camels, the third son  camels and the wise man took his own camel and went away.
camels and the wise man took his own camel and went away.
We are interested in a vector which is uniform on the sphere of radius . Performing averages on the sphere is hard, so we add a camel to the problem by “upgrading” to a spherically symmetric vector  which has a random length. We want to pick a distribution for which the computation
which has a random length. We want to pick a distribution for which the computation  is easy. Fortunately, we already know such a distribution, the Gaussian distribution, for which we already calculated
is easy. Fortunately, we already know such a distribution, the Gaussian distribution, for which we already calculated  .
.
The Gaussian vector and the uniform vector on the sphere are related by
![\[g = \sqrt{\frac{a}{n}} \omega,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a93345cf94749f98e715e42bc796501c_l3.png "Rendered by QuickLaTeX.com")
where  is the squared length of the Gaussian vector . In particular, has the distribution of the sum of
is the squared length of the Gaussian vector . In particular, has the distribution of the sum of  squared Gaussian random variables, which is known as a
squared Gaussian random variables, which is known as a  random variable with degrees of freedom.
random variable with degrees of freedom.
Now, we take the camel back. Compute the variance of  using the chain rule for variance:
using the chain rule for variance:
![\[\Var(g^\top A g) = \mathbb{E}[\Var(g^\top A g \mid a)] + \Var(\mathbb{E}[g^\top A g \mid a]).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e4e5ed235fbf5b2e026bcc58bd23becc_l3.png "Rendered by QuickLaTeX.com")
Here,  and
and ![\mathbb{E}[ \cdot \mid a]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6035d77442675ac2f790cfb5417e1072_l3.png "Rendered by QuickLaTeX.com") denote the conditional variance and conditional expectation with respect to the random variable . The quick and dirty ways of working with these are to treat the random variable “like a constant” with respect to the conditional variance and expectation.
denote the conditional variance and conditional expectation with respect to the random variable . The quick and dirty ways of working with these are to treat the random variable “like a constant” with respect to the conditional variance and expectation.
Plugging in the formula  and treating “like a constant”, we obtain
and treating “like a constant”, we obtain
![\begin{align*}\Var(g^\top A g) &= \mathbb{E}[\Var(a/n \cdot \omega^\top A \omega \mid a)] + \Var(\mathbb{E}[a/n \cdot \omega^\top A \omega \mid a]) \\&=\mathbb{E}[(a/n)^2\Var(\omega^\top A \omega)] + \Var(a/n \cdot \mathbb{E}[\omega^\top A \omega]) \\&= \frac{1}{n^2} \mathbb{E}[a^2] \cdot \Var(\omega^\top A \omega) + \frac{1}{n^2} \Var(a) |\mathbb{E} [\omega^\top A \omega]|^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-be052caddf1d08941925fa0283784635_l3.png "Rendered by QuickLaTeX.com")
As we mentioned, is a random variable with degrees of freedom and ![\mathbb{E}[a^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-53042ba72cdd3991b14b5d4508da6564_l3.png "Rendered by QuickLaTeX.com") and
and  are known quantities that can be looked up:
are known quantities that can be looked up:
![\[\mathbb{E}[a^2] = n(n+2), \quad \Var(a) = 2n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-59ecda0ea21d92e43e3182d5cc5a7217_l3.png "Rendered by QuickLaTeX.com")
We know and ![\mathbb{E} [\omega^\top A \omega] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-293908b9583df45d93349435d76b981a_l3.png "Rendered by QuickLaTeX.com") . Plugging these all in, we get
. Plugging these all in, we get
![\[2\left\|A\right\|_{\rm F}^2 = \frac{n+2}{n} \Var(\omega^\top A\omega) + \frac{2}{n} |\tr A|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c9b70fd4e5bc6ca4006114830d45619_l3.png "Rendered by QuickLaTeX.com")
Rearranging, we obtain
![\[\Var(\omega^\top A\omega) = \frac{2n}{n+2} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n}|\tr A|^2\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7ab53e5895f9a1f474f5bf99fbec5662_l3.png "Rendered by QuickLaTeX.com")
Complex Gaussians
The trick is the same as for real Gaussians. By invariance of complex Gaussian random vectors under unitary transformations, we can reduce to the case where is a diagonal matrix populated with eigenvalues . Then
![\[\Var(\omega^*A \omega) = \Var \left( \sum_{i=1}^n \lambda_i |\omega_i|^2 \right) = \sum_{i=1}^n \Var(|\omega_i|^2) \lambda_i^2 = \sum_{i=1}^n \lambda_i^2 = \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-68f4685a4cd845b98fc1a8fd1494e00a_l3.png "Rendered by QuickLaTeX.com")
Here, we use the fact that  is a random variable with two degrees of freedom, which has variance four.
is a random variable with two degrees of freedom, which has variance four.
Random Phases
The trick is the same as for uniform signs. A short calculation (remembering that is Hermitian and thus  ) reveals that
) reveals that
![\[\Var\left( \omega^* A \omega \right) = \Var \left( \sum_{i<j} 2 \Re(A_{ij} \overline{\omega_i} \omega_j) \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b0e61723c22b056d1aca51694c407e6d_l3.png "Rendered by QuickLaTeX.com")
The random variables  are pairwise independent so we have
are pairwise independent so we have
![\[\Var\left( \omega^* A \omega \right) = \Var \left( \sum_{i<j} 2 \Re(A_{ij} \overline{\omega_i} \omega_j) \right) = 4\sum_{i<j} \Var \left( \Re(A_{ij} \overline{\omega_i} \omega_j) \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f2d77eb554a1e1ffbe4370efd8c704fb_l3.png "Rendered by QuickLaTeX.com")
Since  is uniformly distributed on the complex unit circle, we can assume without loss of generality that
is uniformly distributed on the complex unit circle, we can assume without loss of generality that  . Thus, letting
. Thus, letting  be uniform on the complex unit circle,
be uniform on the complex unit circle,
![\[\Var\left( \omega^* A \omega \right) = 4\sum_{i<j} \Var \left( |A_{ij}|\Re(\phi)) \right) = 4\Var\left( \Re(\phi) \right)\sum_{i<j}|A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-76d2ce84714c4549a86686259bd96847_l3.png "Rendered by QuickLaTeX.com")
The real and imaginary parts of have the same distribution so
![\[1 = \Var(\phi) = \Var(\Re \phi) + \Var(\Im \phi) = 2 \Var(\Re \phi)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f83462ae4de80311482849de445edd5a_l3.png "Rendered by QuickLaTeX.com")
so  . Thus
. Thus
![\[\Var\left( \omega^* A \omega \right) = 2 \sum_{i<j}|A_{ij}|^2 = \sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-436eeb79ad0eddde0752f1a398397ad5_l3.png "Rendered by QuickLaTeX.com")
Uniform on the Complex Sphere: Derivation 1 by Reduction to Real Case
There are at least three simple ways of deriving this result: the camel trick, reduction to the real case, and Haar integration. Each of these techniques illustrates a trick that is useful in its own right beyond the context of trace estimation. Since we have already seen an example of the camel trick for the real sphere, I will present the other two derivations.
Let us begin with the reduction to the real case. Let  and
and  denote the real and imaginary parts of a vector or matrix, taken entrywise. The key insight is that if is a uniform random vector on the complex sphere of radius , then
denote the real and imaginary parts of a vector or matrix, taken entrywise. The key insight is that if is a uniform random vector on the complex sphere of radius , then
![\[\mathscr{R}(\omega) := \twobyone{\mathfrak{R}(\omega)}{\mathfrak{I}(\omega)}\in\real^{2n} \quad \text{is a uniform random vector on the real sphere of radius $\sqrt{n}$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0ff873eff96e863cf02d36e65a5ca1be_l3.png "Rendered by QuickLaTeX.com")
We’ve converted the complex vector into a real vector  .
.
Now, we need to convert the complex matrix into a real matrix  . To do this, recall that one way of representing complex numbers is by
. To do this, recall that one way of representing complex numbers is by  matrices:
matrices:
![\[a + bi \iff \twobytwo{a}{-b}{b}{a}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-21b802836b33431568e5d854455d87d1_l3.png "Rendered by QuickLaTeX.com")
Using this correspondence addition and multiplication of complex numbers can be carried by addition and multiplication of the corresponding matrices.
To convert complex matrices to real matrices, we use a matrix-version of the same representation:
![\[\mathscr{R}(A) = \twobytwo{\mathfrak{R}(A)}{-\mathfrak{I}(A)}{\mathfrak{I}(A)}{\mathfrak{R}(A)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-12a37c4298c9e2d49dfa2f3ac32fe573_l3.png "Rendered by QuickLaTeX.com")
One can check that addition and multiplication of complex matrices can be carried out by addition and multiplication of the corresponding “realified” matrices, i.e.,
![\[\mathscr{R}(A + B) = \mathscr{R}(A) + \mathscr{R}(B), \quad \mathscr{R}(A\cdot B) = \mathscr{R}(A) \cdot \mathscr{R}(B)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4e7fe7da4edb58f1ee1bb8a2b1d2a78b_l3.png "Rendered by QuickLaTeX.com")
holds for all complex matrices and .
We’ve now converted complex matrix and vector into real matrix and vector . Let’s compare to  . A short calculation reveals
. A short calculation reveals
![\[\omega^*A\omega = \mathscr{R}(\omega)^\top \mathscr{R}(A)\mathscr{R}(\omega) .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d92927d567b8907621af83ab283672a5_l3.png "Rendered by QuickLaTeX.com")
Since is a uniform random vector on the sphere of radius ,  is a uniform random vector on the sphere of radius
is a uniform random vector on the sphere of radius  . Thus, by the variance formula for the real sphere, we get
. Thus, by the variance formula for the real sphere, we get
![\[\Var(\omega^*A\omega) = \Var[(\sqrt{2}\mathscr{R}(\omega))^\top (\mathscr{R}(A)/2)(\sqrt{2}\mathscr{R}(\omega) )] = \frac{4n}{2n+2} \left[ \|\mathscr{R}(A)/2\|_{\rm F}^2 - \frac{1}{8n}(\tr\mathscr{R}(A))^2 \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff16ba8dd015218ea642633d77f2a37a_l3.png "Rendered by QuickLaTeX.com")
A short calculation verifies that  and
and  . Plugging this in, we obtain
. Plugging this in, we obtain
![\[\Var(\omega^*A\omega)= \frac{n}{n+1} \left[ \|A\|_{\rm F}^2 - \frac{1}{n}(\tr A)^2 \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c9186eb813af3cd67173d73e32885a22_l3.png "Rendered by QuickLaTeX.com")
Uniform on the Complex Sphere: Derivation 2 by Haar Integration
The proof by reduction to the real case requires some cumbersome calculations and requires that we have already computed the variance in the real case by some other means. The method of Haar integration is more slick, but it requires some pretty high-power machinery. Haar integration may be a little bit overkill for this problem, but this technique is worth learning as it can handle some truly nasty expected value computations that appear, for example, in quantum information.
We seek to compute
![\[\mathbb{E} [(\omega^*A \omega)^2].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8e3e10402f389b06dcd42db97c5ed116_l3.png "Rendered by QuickLaTeX.com")
The first trick will be to write this expession using a single matrix trace using the tensor (Kronecker) product  . For those unfamiliar with the tensor product, the main properties we will be using are
. For those unfamiliar with the tensor product, the main properties we will be using are
(6) ![\[(A\otimes B) (C\otimes D) = (AB) \otimes (CD), \quad \tr(A\otimes B) = \tr A \cdot \tr B. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c5ea1a0235766ad15b67d02b47cfea2e_l3.png "Rendered by QuickLaTeX.com")
We saw in the proof of unbiasedness that
![\[\omega^* A \omega = \tr (\omega^*A\omega) = \tr (A \omega\omega^*).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-578bb521ecfb65fc4025f13431673b08_l3.png "Rendered by QuickLaTeX.com")
Therefore, by (6),
![\[(\omega^*A\omega)^2 = (\tr [A \omega\omega^*])^2 = \tr [A\omega\omega^* \otimes A\omega\omega^*] = \tr [(A\otimes A) (\omega\omega^* \otimes \omega\omega^*)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5161bc0d787be08130c4cfaed0c84489_l3.png "Rendered by QuickLaTeX.com")
Thus, to evaluate ![\mathbb{E}[(\omega^*A\omega)^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-98fe9a5e8220c22709c9d6ade6c3141b_l3.png "Rendered by QuickLaTeX.com") , it will be sufficient to evaluate
, it will be sufficient to evaluate ![\mathbb{E}[\omega\omega^* \otimes \omega\omega^*]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06ac3d64041068176a86563cec4c67ba_l3.png "Rendered by QuickLaTeX.com") . Forunately, there is a useful formula for these expectation provided by a field of mathematics known as representation theory (see Lemma 1 in this paper):
. Forunately, there is a useful formula for these expectation provided by a field of mathematics known as representation theory (see Lemma 1 in this paper):
![\[\mathbb{E}[ \omega\omega^* \otimes \omega\omega^*] = \frac{2n}{n+1} \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e5eddca3275c6bbb82fa6b789efe7664_l3.png "Rendered by QuickLaTeX.com")
Here,  is the orthogonal projection onto the space of symmetric two-tensors
is the orthogonal projection onto the space of symmetric two-tensors  . Therefore, we have that
. Therefore, we have that
![\[\mathbb{E}[(\omega^*A\omega)^2] = \tr [(A\otimes A) \mathbb{E}(\omega\omega^* \otimes \omega\omega^*)] = \frac{2n}{n+1} \tr [(A\otimes A) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6c4f09e537808effd6c51c0aa4d20f6e_l3.png "Rendered by QuickLaTeX.com")
To evalute the trace on the right-hand side of this equation, there is another formula (see Lemma 6 in this paper):
![\[\tr \left[(A\otimes B) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}\right] = \frac{1}{2} \left( \tr(AB) + \tr A \cdot \tr B \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3ea7660d7e72964cf11ef2239731a497_l3.png "Rendered by QuickLaTeX.com")
Therefore, we conclude
![\begin{align*}\Var(\omega^* A \omega) &= \mathbb{E}[(\omega^*A\omega)^2] - (\mathbb{E}[\omega^*A\omega])^2 \\&= \frac{2n}{n+1}\tr [(A\otimes A) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}] - (\tr A)^2 \\&= \frac{n}{n+1}\left[ \tr A^2 + (\tr A)^2 \right] - (\tr A)^2 \\&= \frac{n}{n+1}\left[ \left\|A\right\|_{\rm F}^2 - \frac{1}{n} (\tr A)^2 \right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7af313e9e07f20b93b0de83867f20a04_l3.png "Rendered by QuickLaTeX.com")
Proof of Optimality Properties
In this section, we provide proofs of the two optimality properties.
Optimality: Independent Vectors with Independent Coordinates
Assume is real and symmetric and suppose that is isotropic (2) with independent coordinates. The isotropy condition
![\[\mathbb{E}[\omega\omega^\top] = I\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-232c1e956304b14a7a6f4b55f9ba35e3_l3.png "Rendered by QuickLaTeX.com")
implies that ![\mathbb{E}[\omega_i\omega_j] = \delta_{ij}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9986528214c0653217367aca2a90d568_l3.png "Rendered by QuickLaTeX.com") , where
, where  is the Kronecker symbol. Using this fact, we compute the second moment:
is the Kronecker symbol. Using this fact, we compute the second moment:
![\begin{align*}\mathbb{E}[ (\omega^*A \omega)^2] &= \mathbb{E}\left[ \left( \sum_{i=1}^n A_{ii} \omega_i^2 +2 \sum_{i<j} A_{ij}\omega_i\omega_j) \right)^2\right] \\&= \sum_{i=1}^n A_{ii}^2 \mathbb{E}[\omega_i^4] + \sum_{i<j} (2A_{ii}A_{jj}+4A_{ij}^2) \mathbb{E}[\omega_i^2]\mathbb{E}[\omega_j^2] \\&= \sum_{i=1}^n A_{ii}^2 \mathbb{E}[\omega_i^4] + \sum_{i<j} (2A_{ii}A_{jj}+4A_{ij}^2) .\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4115914a6e648438e66028437f38ef2a_l3.png "Rendered by QuickLaTeX.com")
Thus
![\[\Var(\omega^*A\omega) = \mathbb{E}[ (\omega^*A \omega)^2] - (\mathbb{E}[\omega^* A \omega])^2 = \sum_{i=1}^n A_{ii}^2 (\mathbb{E}[|\omega_i|^4]-1) + 4\sum_{i<j} A_{ij}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-006e2eb8d5f2162654d8c2de39f5a8bc_l3.png "Rendered by QuickLaTeX.com")
The variance is minimized by choosing with  as small as possible. Since
as small as possible. Since  , the smallest possible value for is
, the smallest possible value for is  , which is obtained by populating with random signs.
, which is obtained by populating with random signs.
Optimality: Independent Vectors
This result appears to have first been proven by Richard Kueng in unpublished work. We use an argument suggested to me by Robert J. Webber.
Assume is a class of -Hermitian matrices closed under -unitary similarity transformations and that is an isotropic random vector (2). Decompose the test vector as
![\[\omega = a \cdot s \quad \text{for} \quad a \in [0,+\infty), \: s \in\{x\in \field^n : x^*x = n \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-11867b7bff40d97d79a74d5136774fd9_l3.png "Rendered by QuickLaTeX.com")
First, we shall show that the variance is reduced by replacing  with a vector
with a vector  drawn uniformly from the sphere
drawn uniformly from the sphere
(7) ![\[\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega}) \le \sup_{A\in\mathscr{A}} \Var(\omega^*A\omega \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e54c170954072838232ab5ff7c78cf25_l3.png "Rendered by QuickLaTeX.com")
where
(8) ![\[\tilde{\omega} = a\cdot t \quad \text{and}\quad t\sim \text{Uniform} \{ x \in \field^n :x^*x = n \} \quad \text{is independent of $a$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cb668b2c9f168bca81deb51a17726b1d_l3.png "Rendered by QuickLaTeX.com")
Note that such a can be generated as  for a uniformly random -unitary matrix . Therefore, we have
for a uniformly random -unitary matrix . Therefore, we have
![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[(\tilde{\omega}^*A\tilde{\omega})^2] - (\tr A)^2\right]\\&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-83564c7ba654655b42890c8adb65636f_l3.png "Rendered by QuickLaTeX.com")
Now apply Jensen’s inequality only over the randomness in to obtain
![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right] \\&\le \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f182f68bcb3cbe40c8b44cf5b585e474_l3.png "Rendered by QuickLaTeX.com")
Finally, note that since is closed under -unitary similarity transformations, the supremum over  for
for  is the same as the supremum of , so we obtain
is the same as the supremum of , so we obtain
![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&\le \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right] \\&= \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*As] - (\tr A)^2\right] \\&= \sup_{A\in\mathscr{A}} \Var(\omega^*A\omega).\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bd5736cb133d33ed28ab0f2305ac9678_l3.png "Rendered by QuickLaTeX.com")
We have successfully proven (7). This argument is a specialized version of a far more general result which appears as Proposition 4.1 in this paper.
Next, we shall prove
(9) ![\[\sup_{A\in\mathscr{A}} \Var(t^*At) \le \sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega}), \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0de32ccbfe09388b9d36757b7f140a8e_l3.png "Rendered by QuickLaTeX.com")
where is still defined as in (8). Indeed, using the chain rule for variance, we obtain
![\begin{align*}\Var(\tilde{\omega}^*A\tilde{\omega})&= \Var(a^2\cdot t^*At) \\&= \mathbb{E}[\Var(a^2\cdot t^* A t \mid a)] + \Var(\mathbb{E}[a^2\cdot t^* A t \mid a]) \\&= \mathbb{E}[a^4]\Var(t^* A t )+ (\tr A)^2\Var(a^2) \\&\ge \mathbb{E}[a^4]\Var(t^* A t ).\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c74465c627f5e70f943ce415fbfafd22_l3.png "Rendered by QuickLaTeX.com")
Here, we have used that is uniform on the sphere and thus ![\mathbb{E}[t^*At] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6ee9ad07ec8316cc6003116e3c311189_l3.png "Rendered by QuickLaTeX.com") . By definition, is the length of divided by . Therefore,
. By definition, is the length of divided by . Therefore,
![\[\mathbb{E}[a^2] = \frac{1}{n}\mathbb{E}[\omega^*\omega] = \frac{1}{n} \mathbb{E}[\tr (\omega\omega^*)] = \frac{1}{n} \tr (\mathbb{E}[\omega\omega^*]) = \frac{\tr I}{n} = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4da8a9a33ffb6559da8625435549f7c2_l3.png "Rendered by QuickLaTeX.com")
Therefore, by Jensen’s inequality,
![\[\mathbb{E}[a^4] = \mathbb{E}[(a^2)^2] \ge (\mathbb{E}[a^2])^2 = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-356fd56d5d04dbda45fa992ae083be0f_l3.png "Rendered by QuickLaTeX.com")
Thus
![\[\Var(\tilde{\omega}^*A\tilde{\omega}) \ge \mathbb{E}[a^4]\Var(t^* A t ) \ge \Var(t^*At) \quad \text{for every }A,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fc9331e3508e9102014ecc83f4517cc5_l3.png "Rendered by QuickLaTeX.com")
which proves (9).
 matrix
matrix  matrix, perform the following steps:
matrix, perform the following steps: where
where  is an appropriately chosen
is an appropriately chosen  random matrix.
random matrix. by, e.g., thin QR factorization.
by, e.g., thin QR factorization. . (Here,
. (Here, ![\[B \approx X \coloneqq Q C.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c5eb6a2bd93f7c628f485bd473e8676c_l3.png "Rendered by QuickLaTeX.com")
 to a compact SVD format:
to a compact SVD format: where
where  and
and  and
and  and
and  is
is  .
. in compact SVD form:
in compact SVD form:![\[B \approx X = QC = U\Sigma V^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8f7c31a46a04e9f0447ddc9209f26458_l3.png "Rendered by QuickLaTeX.com")
 ,
,  as estimates of the singular vectors and values of the matrix
as estimates of the singular vectors and values of the matrix  numbers of storage, whereas
numbers of storage, whereas  numbers of storage. As I detailed at length in my blog post on low-rank matrices, many operations are cheap for the low-rank matrix
numbers of storage. As I detailed at length in my blog post on low-rank matrices, many operations are cheap for the low-rank matrix  in roughly
in roughly  . For these use cases, we don’t need the “SVD” part of the randomized SVD.
. For these use cases, we don’t need the “SVD” part of the randomized SVD. to the matrix
to the matrix  is than
is than  .
.![\[\mathbb{E} \left\|B - X\right\|_{\rm F}^2 \le \min_{r \le k-2} \left( 1 + \frac{r}{k-(r+1)} \right) \left\|B - \lowrank{B}_r\right\|^2_{\rm F}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-aa4c5d7163bac127432706a279ae7210_l3.png "Rendered by QuickLaTeX.com")
 approximation error for any
approximation error for any  . In particular, choosing
. In particular, choosing  , we see that the randomized SVD error is at most
, we see that the randomized SVD error is at most  times the best rank-
times the best rank-![\[\mathbb{E} \left\|B - X\right\|_{\rm F}^2 \le (1+r) \left\|B - \lowrank{B}_r \right\|^2_{\rm F} \quad \text{for $k=r+2$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-29753ca17ed5f7def9297cf52157afb8_l3.png "Rendered by QuickLaTeX.com")
 , we see that the randomized SVD has at most twice the error of the best rank-
, we see that the randomized SVD has at most twice the error of the best rank-![\[\mathbb{E} \left\|B - X\right\|_{\rm F}^2 \le 2 \left\|B - \lowrank{B}_r\right\|^2_{\rm F} \quad \text{for $k=2r+1$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c11cf4647747653a356178b65c3585f_l3.png "Rendered by QuickLaTeX.com")
 for the randomized SVD suffices. These results hold even for worst-case matrices. For nice matrices with steadily decaying singular values, the randomized SVD can perform even better than equations (2)–(3) would suggest.
for the randomized SVD suffices. These results hold even for worst-case matrices. For nice matrices with steadily decaying singular values, the randomized SVD can perform even better than equations (2)–(3) would suggest.![\[\mathbb{E} \left\|B - X\right\|^2 \le \min_{r \le k-2} \left( 1 + \frac{2r}{k-(r+1)} \right) \left(\left\|B - \lowrank{B}_r \right\|^2 + \frac{\mathrm{e}^2}{k-r} \left\|B - \lowrank{B}_r\right\|^2_{\rm F} \right). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8ee6f467b55a9e949f3636deca2589a5_l3.png "Rendered by QuickLaTeX.com")
 .
.![\[\norm{B} = \sigma_1(B),\quad \left\|B\right\|_{\rm F}^2 = \sum_i \sigma_i(B)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d3f6deda657f9f0f863a05450571e23_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} \left\|B - X\right\|^2 \le \min_{r \le k-2} \left( 1 + \frac{2r}{k-(r+1)} \right) \left(\sigma_{r+1}^2 + \frac{\mathrm{e}^2}{k-r} \sum_{i>r} \sigma_i^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d4bb8b98ee0928e647c7cdbd91332fcc_l3.png "Rendered by QuickLaTeX.com")
 , this bound demonstrates that the randomized SVD incurs errors based on the entire tail of
, this bound demonstrates that the randomized SVD incurs errors based on the entire tail of  . The randomized SVD is much worse than the best rank-
. The randomized SVD is much worse than the best rank- . Powering has the effect of amplifying the large singular values of
. Powering has the effect of amplifying the large singular values of  we compute the block Krylov sequence
we compute the block Krylov sequence ![\[B\Omega, BB^*B\Omega, BB^*BB^*B\Omega,\ldots,B(B^*B)^q\Omega\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2584aa789abb8f623fbbb14e114a5461_l3.png "Rendered by QuickLaTeX.com")
![\[Y = \begin{bmatrix} B\Omega & BB^*B\Omega & BB^*BB^*B\Omega& \cdots & B(B^*B)^q\Omega\end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4139403e46250e757c4b558a38b9fba7_l3.png "Rendered by QuickLaTeX.com")
 to obtain accurate results.
to obtain accurate results. a
a  and
and  are defined. Then
are defined. Then ![f(\mathbb{E}X) \le \mathbb{E} [f(X)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-be01f50345229c7ba054de409859eec8_l3.png "Rendered by QuickLaTeX.com") .
. , there exists a slope
, there exists a slope  such that
such that  for all
for all  . Invoke this result at
. Invoke this result at  and
and  and take expectations to conclude that
and take expectations to conclude that![\[\mathbb{E}[m(X - \mathbb{E}X) + f(\mathbb{E}X)] = f(\mathbb{E}X) \le \mathbb{E} [f(X)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c162fcb6103e44c65d3c6e1d003135d0_l3.png "Rendered by QuickLaTeX.com")
 for two numbers
for two numbers  and
and  . This may hard to do directly. With the interpolation method, I first construct a family of numbers
. This may hard to do directly. With the interpolation method, I first construct a family of numbers  ,
,  , such that
, such that  and
and  and show that
and show that  is (weakly) increasing in
is (weakly) increasing in ![\[\frac{d}{dt} A_t \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7d87bd05d1a5b326f575b2b72f27cc74_l3.png "Rendered by QuickLaTeX.com")
 for some
for some  for any Gaussian random variable
for any Gaussian random variable  .
.![\[f(0) \le \mathbb{E} [f(X)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8e5dd3a04978ca82dd4d50fd41f8ca43_l3.png "Rendered by QuickLaTeX.com")
![\[A_t = \mathbb{E} [ f(X_t) ],\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8f12251961aff234ee138a29978a264b_l3.png "Rendered by QuickLaTeX.com")
 starts with no randomness and
starts with no randomness and  is our standard Gaussian. To interpolate between these extremes, we increase the variance linearly from
is our standard Gaussian. To interpolate between these extremes, we increase the variance linearly from  to
to ![\[A_t = \mathbb{E} [ f(X_t)] \quad \text{where $X_t\sim\mathcal{N}(0,t)$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-88cc0a1a3f5c52e8349fd6c4ca28ad7c_l3.png "Rendered by QuickLaTeX.com")
 denotes a Gaussian random variable with zero mean and variance
denotes a Gaussian random variable with zero mean and variance  .
. denote a small parameter which we will later send to zero. For us, the key fact will be that a
denote a small parameter which we will later send to zero. For us, the key fact will be that a  can be realized as a sum of independent
can be realized as a sum of independent  and
and  random variables. Therefore, we write
random variables. Therefore, we write![\[X_{t+\delta} = X_t + \Delta \quad \text{where $\Delta \sim \mathcal{N}(0,\delta)$ is independent of $X_t$.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eaaa1bd888269b9ad51b88afb5fc3d08_l3.png "Rendered by QuickLaTeX.com")
 by using Taylor’s formula
by using Taylor’s formula![\[f(X_t+\Delta) = f(X_t) + f'(X_t)\Delta + \frac{1}{2} f''(X_t) \Delta^2 + \frac{1}{6} f'''(\xi) \Delta^3, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-38d38dde93cc24a3101649064d5af389_l3.png "Rendered by QuickLaTeX.com")
 lies between
lies between  and
and  . Now, take expectations,
. Now, take expectations, ![\[\mathbb{E}[ f(X_t+\Delta)]=\mathbb{E}[f(X_t)] + \mathbb{E}[f'(X_t)\Delta] + \frac{1}{2} \mathbb{E}[f''(X_t)] \mathbb{E}[\Delta^2] + \underbrace{\frac{1}{6} \mathbb{E}[f'''(\xi) \Delta^3]}_{:=\mathrm{Rem}(\delta)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ccacfa2622a31446e66624a9a8458dc4_l3.png "Rendered by QuickLaTeX.com")
 has mean zero and variance
has mean zero and variance ![\[\mathbb{E} [f(X_t+\Delta)]=\mathbb{E}[f(X_t)] + \delta \frac{1}{2} \mathbb{E}[f''(X_t)] + \mathrm{Rem}(\delta).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d021f960b2367c41cf18dc25661045c6_l3.png "Rendered by QuickLaTeX.com")
 vanishes as
vanishes as  . Thus, we can rearrange this expression to compute the derivative:
. Thus, we can rearrange this expression to compute the derivative:![\[\frac{d}{dt} A_t = \lim_{\delta \downarrow 0} \frac{\mathbb{E} f(X_t+\Delta)-\mathbb{E}[f(X_t)]}{\delta} = \lim_{\delta \downarrow 0} \frac{1}{2} \mathbb{E}[f''(X_t)] + \frac{\mathrm{Rem}(\delta)}{\delta} = \frac{1}{2} \mathbb{E}[f''(X_t)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3275fd914e25d0f6985eb4d1ca419fb7_l3.png "Rendered by QuickLaTeX.com")
 for every
for every  . Therefore,
. Therefore,![\[\frac{d}{dt} A_t \ge 0 \quad \text{for all } t\in [0,1].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-258c784d449d2b53c0a462ff38f3cdd8_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} f(X) = f(0) + \frac{1}{2} \int_0^1 \mathbb{E} [f''(X_t)] \, dt.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-50be5d67c30ff436025f45185c2fae22_l3.png "Rendered by QuickLaTeX.com")
 is
is 
![\[f''(x) \le \beta \quad \text{for every } x \in \real,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-206d02bd0b654ab4c42cf33921d5383d_l3.png "Rendered by QuickLaTeX.com")
![\[f(0) \le \mathbb{E} f(X) \le f(0) + \frac{1}{2}\beta.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8561680db386f740b927a84c7cb9fe0d_l3.png "Rendered by QuickLaTeX.com")
 . Let’s do this. As an exercise, you can verify that our technical regularity condition implies
. Let’s do this. As an exercise, you can verify that our technical regularity condition implies  . Thus, by Hölder’s inequality and setting
. Thus, by Hölder’s inequality and setting  ‘s Hölder conjugate (
‘s Hölder conjugate ( ), we obtain
), we obtain![\[\frac{|\mathrm{Rem}(\delta)|}{\delta} = \frac{|\mathbb{E}[f'''(\xi) \Delta^3]|}{6\delta} \le \frac{(|\mathbb{E} |f'''(\xi)|^p)^{1/p}| (\mathbb{E} |\Delta|^{3q})^{1/q}}{6\delta}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e1078e0f2d2c17b77bd87a38665c03e4_l3.png "Rendered by QuickLaTeX.com")
 where
where  is a function of
is a function of  as
as  .
. , defined for each
, defined for each  . Rather than treating these quantities as independent, we can think of them as a collective, comprising a random function
. Rather than treating these quantities as independent, we can think of them as a collective, comprising a random function  known as a
known as a 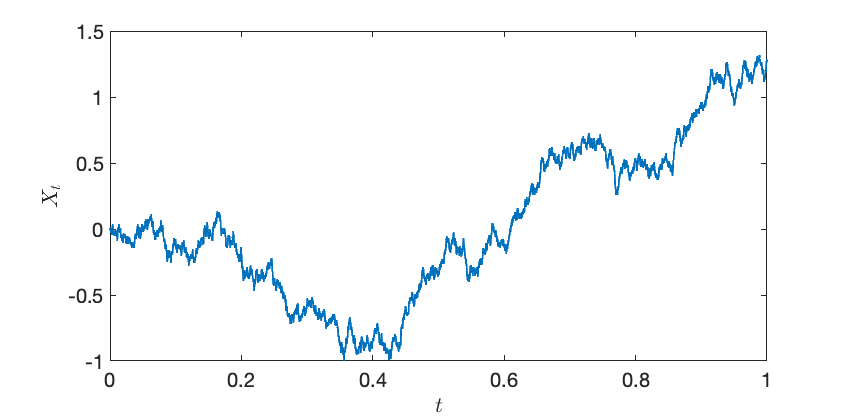
 and a Gaussian with mean
and a Gaussian with mean ![\[df = f'(x) \, dx\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a32870f7f5981a412b96de2591534280_l3.png "Rendered by QuickLaTeX.com")
 of a Brownian motion, the analog is
of a Brownian motion, the analog is ![\[df = f'(X_t) \, dX_t + \frac{1}{2} f''(X_t) \, dt.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e2eadd9f0624f41b562d90fe0568ef69_l3.png "Rendered by QuickLaTeX.com")
 over a time interval
over a time interval  ,
,  is a random variable with mean
is a random variable with mean ![\[f(\mathbb{E}Y) \le \mathbb{E}[f(Y)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-97693077e012820448760d2e7ad31d58_l3.png "Rendered by QuickLaTeX.com")
 and
and  denote the cumulative distribution functions of
denote the cumulative distribution functions of ![\[g(X) := \inf \{ \alpha \in \real : F_Y(\alpha) \ge F_X(X) \}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2c40cdc3f3bcf589f930aeb87340491b_l3.png "Rendered by QuickLaTeX.com")
 and
and ![\[A_t \stackrel{?}{=} \mathbb{E} [f(g(X_t))].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0de3d1811026212f153ea79469649149_l3.png "Rendered by QuickLaTeX.com")
![A_0 = \mathbb{E}[f(g(0))]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e6ac6421e3fc94f2087614f41336491a_l3.png "Rendered by QuickLaTeX.com") does not even equal to
does not even equal to ![\mathbb{E} [f(Y)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06983640d9e119e9aa44cdd209c540c7_l3.png "Rendered by QuickLaTeX.com") ! Instead, we must define
! Instead, we must define![\[A_t = \mathbb{E} [f(\mathbb{E}[g(X_1) \mid X_t])].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-19560c3c89aa3987101e6650af24a0a7_l3.png "Rendered by QuickLaTeX.com")
 conditional on the Brownian motion
conditional on the Brownian motion ![\[\frac{d}{dt} A_t \ge 0 \quad \text{for all }t\in [0,1].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-253003b644ae67d253cdfd74378c71a9_l3.png "Rendered by QuickLaTeX.com")
 are “somewhat dependent”, for which functions does the multivariate Jensen’s inequality
are “somewhat dependent”, for which functions does the multivariate Jensen’s inequality  )
) ![\[f(\mathbb{E} Y_1,\ldots,\mathbb{E}Y_n) \le \mathbb{E} [f(Y_1,\ldots,Y_n)] \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2467fe840ba04173b1a3adc7c38a945c_l3.png "Rendered by QuickLaTeX.com")
![\[\text{($\star$) holds if $f$ is convex in each coordinate, separately.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-521f389e0e1a83728c2544ecb889a047_l3.png "Rendered by QuickLaTeX.com")
 of Gaussian random variables
of Gaussian random variables  . We will use the covariance matrix
. We will use the covariance matrix ![\[f(\mathbb{E}g_1(X_1),\ldots,\mathbb{E}g_n(X_n)) \le \mathbb{E} [f(g(X_1),\ldots,g(X_n))]\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-055c60480c4de2b28348bb1da12763ea_l3.png "Rendered by QuickLaTeX.com")
 if and only if
if and only if ![\[\Sigma \circ \nabla^2 f(x) \text{ is positive semidefinite} \quad \text{for all $x \in \real^n$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-48ca7d18e3bbcee337d072bd27efe791_l3.png "Rendered by QuickLaTeX.com")
 is the
is the  denotes the
denotes the  are equal (and variance one),
are equal (and variance one),  for all
for all ![\[\Sigma \circ \nabla^2 f(x) = \mathrm{diag} \left( \frac{\partial^2 f}{\partial x_i^2} : i=1,\ldots,n \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ef80f7d5a452131378d194e3560717b0_l3.png "Rendered by QuickLaTeX.com")
 : this is precisely the condition for
: this is precisely the condition for  . At each step, the user makes one of two choices:
. At each step, the user makes one of two choices:
 .
. is a fixed number
is a fixed number  . In particular, the probability
. In particular, the probability  by
by  . Note that the states
. Note that the states ![\[\mathbb{P} \{ x_{n+1} = j \mid x_n = i,x_{n-1}=a_{n-1},\ldots,x_0=a_0\} = \mathbb{P}\{x_{n+1} = j \mid x_n = i\} = P_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d1fa5c29673402a40f530434ec99f998_l3.png "Rendered by QuickLaTeX.com")
 given the entire history of the system depends only on the value
given the entire history of the system depends only on the value  of the chain at time
of the chain at time  .
. )
) ![\[P_{ij} = \frac{0.15}{m}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e9cb5a0e824c976c19d2d73c1f45714b_l3.png "Rendered by QuickLaTeX.com")
 outgoing links. Then, in addition to the
outgoing links. Then, in addition to the  probability computed before, user
probability computed before, user  chance of picking
chance of picking  )
) ![\[P_{ij} = \frac{0.85}{d_i} + \frac{0.15}{m}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-51a6f3c00da58822f6465db8458a1f8c_l3.png "Rendered by QuickLaTeX.com")
 , we can understand the processes evolution by determining its state
, we can understand the processes evolution by determining its state  at every point in time
at every point in time  of the process at every time
of the process at every time  by a row vector
by a row vector  .
. is a column vector but
is a column vector but  stores the probability that the system is in state
stores the probability that the system is in state  for every
for every  ).
). denote the probability distributions of the states
denote the probability distributions of the states  . It is natural to ask: How are the distributions
. It is natural to ask: How are the distributions  is in state
is in state  :
:![\[\rho^{(n+1)}_j = \mathbb{P} \{x_{n+1} = j\}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d8705a90b65f869a58cd432bbc884cfa_l3.png "Rendered by QuickLaTeX.com")
 or
or  or … or
or … or  ; only one of these cases can be true at once. When we have an “or” of random events and these events are
; only one of these cases can be true at once. When we have an “or” of random events and these events are ![\[\rho^{(n+1)}_j = \mathbb{P} \{x_{n+1} = j\} = \sum_{i=1}^m \mathbb{P} \{x_{n+1} = j, x_n = i\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-448c23761c566dc3504d60b57f1ac8d1_l3.png "Rendered by QuickLaTeX.com")
 and
and ![\[\rho^{(n+1)}_j = \sum_{i=1}^m \mathbb{P} \{x_{n+1} = j, x_n = i\} = \sum_{i=1}^m \mathbb{P} \{x_n = i\} \mathbb{P}\{x_{n+1} = j \mid x_n = i\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9b27f4d81b8b51c36f578f7e00017d96_l3.png "Rendered by QuickLaTeX.com")
 and the probability of moving from
and the probability of moving from ![\[\rho_j^{(n+1)} = \sum_{i=1}^m \rho^{(n)}_i P_{ij} .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1a858376daf68ec5f2d816ef52e66997_l3.png "Rendered by QuickLaTeX.com")
![\[\left(\rho^{(n+1)}\right)^\top = \left(\rho^{(n)}\right)^\top P \quad \text{for any } n = 0,1,2,\ldots.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9ae710d7c26dc66ed497cc8faf34cab3_l3.png "Rendered by QuickLaTeX.com")
 matrix
matrix  , then the distribution at time
, then the distribution at time ![\[\left(\rho^{(n)}\right)^\top = \left(\rho^{(n-1)}\right)^\top P = \left[\left(\rho^{(n-2)}\right)^\top P\right]P = \left(\rho^{(n-2)}\right)^\top P^2 = \cdots = \left(\rho^{(0)}\right)^\top P^n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3fddd821b8cdb4b89262b0c72ea68e55_l3.png "Rendered by QuickLaTeX.com")
 at time
at time  converge to a single fixed probability distribution
converge to a single fixed probability distribution  regardless of how the chain is initialized (i.e., independent of the starting distribution
regardless of how the chain is initialized (i.e., independent of the starting distribution ![\[\pi^\top = \pi^\top P. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-daa1101ffcda26584d5f843c12765158_l3.png "Rendered by QuickLaTeX.com")
![\[\left(\rho^{(n+1)}\right)^\top = \left(\rho^{(n)}\right)^\top P,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bcbc3e101491a613c1dcb40680a4ef3b_l3.png "Rendered by QuickLaTeX.com")
 , and observe that both
, and observe that both  and
and  converge to
converge to  .
.![\[\text{There exists $n$ such that, for any $i,j = 1,2,\ldots,m$, } \quad\mathbb{P}\{x_n = j \mid x_0 = i \} = (P^n)_{ij} > 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9796d227fdde3830779ea16f255b9b70_l3.png "Rendered by QuickLaTeX.com")
 converge to
converge to  step, there is at least a
step, there is at least a  chance of moving from any website
chance of moving from any website  and you’re interested in where your traffic is coming from. One way of achieving this would be to initialize the Markov chain at
and you’re interested in where your traffic is coming from. One way of achieving this would be to initialize the Markov chain at  as our initial distribution for
as our initial distribution for  is defined as follows: The probability
is defined as follows: The probability  of moving from
of moving from ![\[\mathbb{P} \{y_1 = j \mid y_0 = i\} = P^{\rm rev}_{ij} = \mathbb{P} \{ x_0 = j \mid x_1 = i \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-caeb5343a623f571314596e612b7315d_l3.png "Rendered by QuickLaTeX.com")
![\[P^{\rm rev}_{ij} = \mathbb{P} \{ x_0 = j \mid x_1 = i \} = \frac{\mathbb{P} \{x_0 = j\} \mathbb{P} \{x_1 = i \mid x_0 = j\}}{\mathbb{P} \{x_1 = i\}} = \frac{ \pi_j P_{ji}}{\pi_i}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1ffaf13a6eb4a6d8079f1ee0f7501d5d_l3.png "Rendered by QuickLaTeX.com")
 at their website and following the chain one step back.
at their website and following the chain one step back.![\[P^{\rm rev}_{ij} = P_{ij} \quad \text{for every } i,j=1,2,\ldots,m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4d394cf7f8d6b8a6318c27c3e447403d_l3.png "Rendered by QuickLaTeX.com")
![\[\pi_i P_{ij} = \pi_j P_{ji}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d6dade4e26238f165b12b7d2891d3a95_l3.png "Rendered by QuickLaTeX.com")
 as defining a function on the set
as defining a function on the set  ,
,  . Letting
. Letting  , we can define a non-standard
, we can define a non-standard  :
: ![\langle f, g\rangle_{\pi} \coloneqq \mathbb{E}[f(x) g(x)] = \sum_{i=1}^m \pi_i f(i)g(i)](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c871929113a513c08da0e9fa24a8043_l3.png "Rendered by QuickLaTeX.com") . Then the Markov chain is reversible if and only if detailed balance holds if and only if
. Then the Markov chain is reversible if and only if detailed balance holds if and only if  . This more abstract characterization has useful consequences. For instance, by the
. This more abstract characterization has useful consequences. For instance, by the  ) is equal to the flow of probability mass from
) is equal to the flow of probability mass from  ).
). :
:![\[\sigma_i P_{ij} = \sigma_j P_{ji}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f1ef7062af77b1ad2efc526c3db8a63b_l3.png "Rendered by QuickLaTeX.com")
 is the stationary distribution of this chain. To see why, we check the stationarity condition
is the stationary distribution of this chain. To see why, we check the stationarity condition  . Indeed, for every
. Indeed, for every ![\[(\sigma^\top P)_j = \sum_{i=1}^m \sigma_i P_{ij} = \sum_{i=1}^m \sigma_j P_{ji} = \sigma_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-81a7b02ba8de821bb56ecee978ec346d_l3.png "Rendered by QuickLaTeX.com")
 such that
such that  for every
for every  . The proposal distribution
. The proposal distribution  add to one:
add to one:  .
. , then
, then  .
.![\[\min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fb8ef7791b1d955015f1300e0eddfb8c_l3.png "Rendered by QuickLaTeX.com")
 . This Markov chain is known as a
. This Markov chain is known as a  .
. with from the proposal distribution,
with from the proposal distribution,  .
.![\[p_{\rm acc} := \min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e34abfe410068f7516cd1c5e50100c3c_l3.png "Rendered by QuickLaTeX.com")
 , set
, set  . Otherwise, set
. Otherwise, set  .
. and go back to step 2.
and go back to step 2. under the Metropolis–Hastings sampler is the proposal probability
under the Metropolis–Hastings sampler is the proposal probability ![\[P_{ij} = T_{ij} \cdot \min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8c98e72078f3de5c0de43a1683f5d406_l3.png "Rendered by QuickLaTeX.com")
 is always satisfied for any Markov chain
is always satisfied for any Markov chain  .
.![\[\pi_i P_{ij} = \pi_i T_{ij} \cdot \min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\} = \min \left\{ \pi_i T_{ij} , \pi_j T_{ji} \right\} = \pi_j P_{ji}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9ec53c4f48f0cc7bb8cc1d0cd58750f1_l3.png "Rendered by QuickLaTeX.com")
 different baked goods. I want to pick out
different baked goods. I want to pick out  th entry
th entry  of my matrix, I write the number of sales for baked good
of my matrix, I write the number of sales for baked good  of my matrix with a measure of similarity between items
of my matrix with a measure of similarity between items  be the number ordered for each bakery item by a random customer. Set
be the number ordered for each bakery item by a random customer. Set  being the correlation between the random variables
being the correlation between the random variables  and
and  . The matrix
. The matrix  is the total sales of item
is the total sales of item  . By scaling
. By scaling  of picking items
of picking items  is proportional to the
is proportional to the  . More specifically,
. More specifically,![\[\pi_S = \frac{\det A(S,S)}{\sum_{\text{all subsets $T$ of size $k$}} \det A(T,T)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-26714c8980fc33f439de8cf1e5a692eb_l3.png "Rendered by QuickLaTeX.com")
 . Such a random subset is known as a
. Such a random subset is known as a  items and a display case of size
items and a display case of size  . Suppose I have three items: a pumpkin muffin, a chocolate chip muffin, and an oatmeal raisin cookies. Say the
. Suppose I have three items: a pumpkin muffin, a chocolate chip muffin, and an oatmeal raisin cookies. Say the ![\[A = \begin{bmatrix} 10 & 9 & 0 \\ 9 & 10 & 0 \\ 0 & 0 & 5 \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8bb20f6bff68d835397eabaf8a1cdc0_l3.png "Rendered by QuickLaTeX.com")
 and much more popular than the cookie
and much more popular than the cookie  . However, the two muffins are similar to each other and thus the corresponding submatrix has small determinant
. However, the two muffins are similar to each other and thus the corresponding submatrix has small determinant![\[\det A(\{1,2\},\{1,2\}) = \det \twobytwo{10}{9}{9}{10} = 19.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7f74f9426bb0c808b9e4384a0a2a6c2d_l3.png "Rendered by QuickLaTeX.com")
![\[\det A(\{1,3\},\{1,3\}) = \det A(\{2,3\},\{2,3\}) = \det \twobytwo{10}{0}{0}{5} = 50.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-812d8318cba1d4a937e92858658ff7e8_l3.png "Rendered by QuickLaTeX.com")
 -DPP, we have a
-DPP, we have a  chance of choosing a muffin and a cookie for our display case. It is for this reason that we can say that a
chance of choosing a muffin and a cookie for our display case. It is for this reason that we can say that a  possible
possible  items and want to pick
items and want to pick  of them, there are already over 10 trillion possible combinations.
of them, there are already over 10 trillion possible combinations. . To generate a proposal, choose a uniformly random element
. To generate a proposal, choose a uniformly random element  out of
out of  and a uniformly random element
and a uniformly random element  out of
out of  without
without  obtained from
obtained from  ).
).![\[p_{\rm acc} = \min \left\{ 1 , \frac{\pi_{S'} T_{S'S}}{\pi_{S} T_{SS'}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5ff5280a20cfddd08b7917152edbfb68_l3.png "Rendered by QuickLaTeX.com")
 and
and  are both equal to
are both equal to  ,
,  . Using the formula for the probability
. Using the formula for the probability ![\[\frac{\pi_{S'}}{\pi_S} = \frac{\det A(S',S')}{\det A(S,S)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-26f4fdbc4d7624e49904e22703e8aaa7_l3.png "Rendered by QuickLaTeX.com")
![\[p_{\rm acc} = \min \left\{ 1 , \frac{\pi_{S'} T_{S'S}}{\pi_{S} T_{SS'}} \right\} = \min \left\{ 1, \frac{\det A(S',S')}{\det A(S,S)} \right\}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-16c835576499e7fb3e6cf2c219732873_l3.png "Rendered by QuickLaTeX.com")
 arbitrarily and set
arbitrarily and set  .
. .
. .
. . Otherwise, set
. Otherwise, set  .
. steps.
steps. entries of the matrix
entries of the matrix  of
of  denotes the
denotes the ![\[\|g\| = \sqrt{g_1^2 + \cdots + g_n^2},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-284e0c747adfdfc57c90b3d282798075_l3.png "Rendered by QuickLaTeX.com")


![\[\mathbb{E} \|g\| = \sqrt{2} \frac{\Gamma((n+1)/2)}{\Gamma(n/2)},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-274737e67b7cf65854623497d2336e99_l3.png "Rendered by QuickLaTeX.com")
 is the
is the ![\[\sqrt{n-1} < \frac{n}{\sqrt{n+1}} < \mathbb{E} \|g\| < \sqrt{n}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-074224f9e909d1c2a33cfadb8b37e771_l3.png "Rendered by QuickLaTeX.com")
 and
and  , we have
, we have![\[\frac{x}{(x+s)^{1-s}} < \frac{\Gamma(x+s)}{\Gamma(x)} < x^s. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-52ca1feab59cc018c9d7af7cbf54afd7_l3.png "Rendered by QuickLaTeX.com")
 and
and  and multiply by
and multiply by  to obtain
to obtain![\[\frac{\sqrt{2} \cdot n/2}{(n/2+1/2)^{1/2}} < \sqrt{2}\frac{\Gamma((n+1)/2)}{\Gamma(n/2)} = \mathbb{E}\|g\| < \sqrt{2}\cdot \sqrt{n/2},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9bbede5bad09ae09b80d77db4e728b62_l3.png "Rendered by QuickLaTeX.com")
 and
and ![\[\Gamma((1-s)x + sy) < \Gamma(x)^{1-s} \Gamma(y)^s. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-53b31e91194bbc5f8b440c65b868feb4_l3.png "Rendered by QuickLaTeX.com")
 to obtain
to obtain![\[\Gamma(x+s) = \Gamma((1-s)x + s(x+1)) < \Gamma(x)^{1-s} \Gamma(x+1)^s.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-00f0550f27637fb27d0ab53fb8892bff_l3.png "Rendered by QuickLaTeX.com")
 and use
and use  to conclude
to conclude![\[\frac{\Gamma(x+s)}{\Gamma(x)} < \left( \frac{\Gamma(x+1)}{\Gamma(x)} \right)^s = x^s. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3607c86e6e40b082b75960abc24dca1c_l3.png "Rendered by QuickLaTeX.com")
 in place of
in place of  in place of
in place of ![\[\frac{\Gamma(x+1)}{\Gamma(x+s)} < (x+s)^{1-s}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6c2671296814cc9b8138d5143fc9d6a9_l3.png "Rendered by QuickLaTeX.com")
 , dividing by
, dividing by  , and using
, and using ![\[\frac{\Gamma(x+s)}{\Gamma(x)} > \frac{\Gamma(x+1)}{\Gamma(x)} \cdot \frac{1}{(x+s)^{1-s}} = \frac{x}{(x+s)^{1-s}},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0d742ab239498d1540847646ffabaa9c_l3.png "Rendered by QuickLaTeX.com")
 , where we use the fact that
, where we use the fact that  is the sum of
is the sum of  follows from a weaker version of Wendel’s inequality,
follows from a weaker version of Wendel’s inequality,  ,
,![\[\Var(f(g)) \le \mathbb{E} \| \nabla f(g)\|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-456ec9d3ca714d36d760fccd6f3a452c_l3.png "Rendered by QuickLaTeX.com")
 which has gradient
which has gradient  . Thus,
. Thus,![\[\mathbb{E} \| \nabla f(g)\|^2 = 1 \ge \Var(f(g)) = \mathbb{E} \|g\|^2 - (\mathbb{E} \|g\|)^2 = n - (\mathbb{E} \|g\|)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b806bd5311f18368939fef93576bf328_l3.png "Rendered by QuickLaTeX.com")
 positive semidefinite (psd) matrix
positive semidefinite (psd) matrix ![\[\hat{A} = A(:,S) \, A(S,S)^{-1} \, A(S,:), \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cf768b8c5fedfd543a62070abae10bfe_l3.png "Rendered by QuickLaTeX.com")
 identifies a subset of
identifies a subset of  , this allowed us to approximate all
, this allowed us to approximate all  entries in columns
entries in columns  of the approximation?
of the approximation? , where
, where  is a vector of length
is a vector of length  of a lower triangular matrix
of a lower triangular matrix  and an upper triangular matrix
and an upper triangular matrix  where
where  for
for  for
for  . This factorization
. This factorization  is known as a
is known as a  , where
, where ![\[A = \twobytwo{A_{11}}{A_{12}}{A_{21}}{A_{22}}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-24b23cd2ac7afc3e7c6acfb7fd8f8220_l3.png "Rendered by QuickLaTeX.com")
 and subtracting this from the second block row introduces a matrix of zeros into the bottom left block of
and subtracting this from the second block row introduces a matrix of zeros into the bottom left block of  is a
is a  for every nonzero vector
for every nonzero vector ![\[\twobytwo{I}{0}{-A_{21}A_{11}^{-1}}{I}\twobytwo{A_{11}}{A_{12}}{A_{21}}{A_{22}} = \twobytwo{A_{11}}{A_{12}}{0}{A_{22} - A_{21}A_{11}^{-1}A_{12}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4b8f6deed9a1dd928c76d311ab88fd54_l3.png "Rendered by QuickLaTeX.com")
![\[\twobytwo{I}{0}{-A_{21}A_{11}^{-1}}{I}^{-1} = \twobytwo{I}{0}{A_{21}A_{11}^{-1}}{I}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-be1610a1241e5c5bfd2c7dc04653e712_l3.png "Rendered by QuickLaTeX.com")
![\[A = \twobytwo{A_{11}}{A_{12}}{A_{21}}{A_{22}} = \twobytwo{I}{0}{A_{21}A_{11}^{-1}}{I} \twobytwo{A_{11}}{A_{12}}{0}{A_{22} - A_{21}A_{11}^{-1}A_{12}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e004e13e06bf86fd8707805ce21f9071_l3.png "Rendered by QuickLaTeX.com")
![\[A = \twobytwo{A_{11}}{A_{12}}{A_{21}}{A_{22}} = \twobytwo{I}{0}{A_{21}A_{11}^{-1}}{I} \twobytwo{A_{11}}{0}{0}{A_{22} - A_{21}A_{11}^{-1}A_{12}} \twobytwo{I}{0}{A_{21}A_{11}^{-1}}{I}^*. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7e3a03d3b1e4421e71d17e2abba7ef97_l3.png "Rendered by QuickLaTeX.com")
 , where
, where ![\[S = A_{22} - A_{21}A_{11}^{-1}A_{12}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-55e08632ed61d20591817565b66a7e09_l3.png "Rendered by QuickLaTeX.com")
 in matrix theory makes it deserving of its special name, the Schur complement. To us for now, the Schur complement is just the matrix appearing in the bottom right corner of our block Cholesky factorization.
in matrix theory makes it deserving of its special name, the Schur complement. To us for now, the Schur complement is just the matrix appearing in the bottom right corner of our block Cholesky factorization. is positive (semi)definite, then the Schur complement
is positive (semi)definite, then the Schur complement  is positive (semi)definite.
is positive (semi)definite. .
.![\[A_{11} = L_{11}^{\vphantom{*}}L_{11}^*, \quad S = L_{22}^{\vphantom{*}}L_{22}^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fcde81f118ea8516883b47d07a35b2b0_l3.png "Rendered by QuickLaTeX.com")
 factorization (1) and simplifying gives a Cholesky factorization, as desired:
factorization (1) and simplifying gives a Cholesky factorization, as desired:![\[A = \twobytwo{L_{11}}{0}{A_{21}^{\vphantom{*}}(L_{11}^{*})^{-1}}{L_{22}}\twobytwo{L_{11}}{0}{A_{21}^{\vphantom{*}}(L_{11}^{*})^{-1}}{L_{22}}^* =: LL^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e22f8c5665f2154819e960cd6c6b32d3_l3.png "Rendered by QuickLaTeX.com")
 , perform the following steps:
, perform the following steps:![\[L(j:N,j) \leftarrow A(j:N,j)/\sqrt{a_{jj}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9d8a1752f0ae4613f71767ce87b9560a_l3.png "Rendered by QuickLaTeX.com")
![\[A(j+1:N,j+1:N)\leftarrow A(j+1:N,j+1:N) - \frac{A(j+1:N,j)A(j,j+1:N)}{a_{jj}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d8090111efb4d1beb891cac29c04a598_l3.png "Rendered by QuickLaTeX.com")
![\[A = \twobytwo{I}{0}{A_{21}A_{11}^{-1}}{I} \twobytwo{A_{11}}{0}{0}{A_{22} - A_{21}A_{11}^{-1}A_{12}} \twobytwo{I}{0}{A_{21}A_{11}^{-1}}{I}^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7d369f0415a2b62af274f183e8828853_l3.png "Rendered by QuickLaTeX.com")
 , which is a large matrix of size
, which is a large matrix of size  .
.![\[A = \twobyone{I}{A_{21}{A_{11}^{-1}}} A_{11} \twobyone{I}{A_{22}{A_{11}^{-1}}}^* + \twobytwo{0}{0}{0}{A_{22}-A_{21}A_{11}^{-1}A_{12}}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eb7264120d2157941336da3dcbbcc1d8_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{A} = \twobyone{A_{11}}{A_{21}} A_{11}^{-1} \onebytwo{A_{11}}{A_{12}} = \twobytwo{A_{11}}{A_{12}}{A_{21}}{A_{21}A_{11}^{-1}A_{12}},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8ec34b04edefd156d8de3711608998b2_l3.png "Rendered by QuickLaTeX.com")
 and is the final of our three titular characters. The residual of the Nyström approximation is the second term in (2), which is none other than the Schur complement (Sch), padded by rows and columns of zeros:
and is the final of our three titular characters. The residual of the Nyström approximation is the second term in (2), which is none other than the Schur complement (Sch), padded by rows and columns of zeros:![\[A - \hat{A} = \twobytwo{0}{0}{0}{A_{22}-A_{21}A_{11}^{-1}A_{12}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8dccb2f8fd446335886ec9bdabc85da_l3.png "Rendered by QuickLaTeX.com")
 is obtained from the process of terminating a Cholesky factorization midway through algorithm execution, so we say that the Nyström approximation results from a partial Cholesky factorization of the matrix
is obtained from the process of terminating a Cholesky factorization midway through algorithm execution, so we say that the Nyström approximation results from a partial Cholesky factorization of the matrix  followed by position
followed by position  and so on. There’s no need to insist on this exact ordering of elimination steps. Indeed, at each step of the Cholesky algorithm, we can choose whichever diagonal position
and so on. There’s no need to insist on this exact ordering of elimination steps. Indeed, at each step of the Cholesky algorithm, we can choose whichever diagonal position  that we want to perform elimination. The entry we choose to perform elimination with is called a
that we want to perform elimination. The entry we choose to perform elimination with is called a  matrix
matrix  to store the column Nyström approximation
to store the column Nyström approximation  , in factored form. For
, in factored form. For  , perform the following steps:
, perform the following steps: .
. .
. .
.![\[\hat{A} = FF^* = A(:,S) \, A(S,S)^{-1} \, A(S,:).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f9ac31fa249d889cd909be1d340abd58_l3.png "Rendered by QuickLaTeX.com")
![\[s_j = \argmax_{1\le k\le N} a_{kk}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-42a58dd2bd6824be5efdc930fc5356df_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ s_j = k \} = \frac{a_{kk}}{\operatorname{tr} A}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-36ca3921c45ffb034e0cfa8adf85b982_l3.png "Rendered by QuickLaTeX.com")
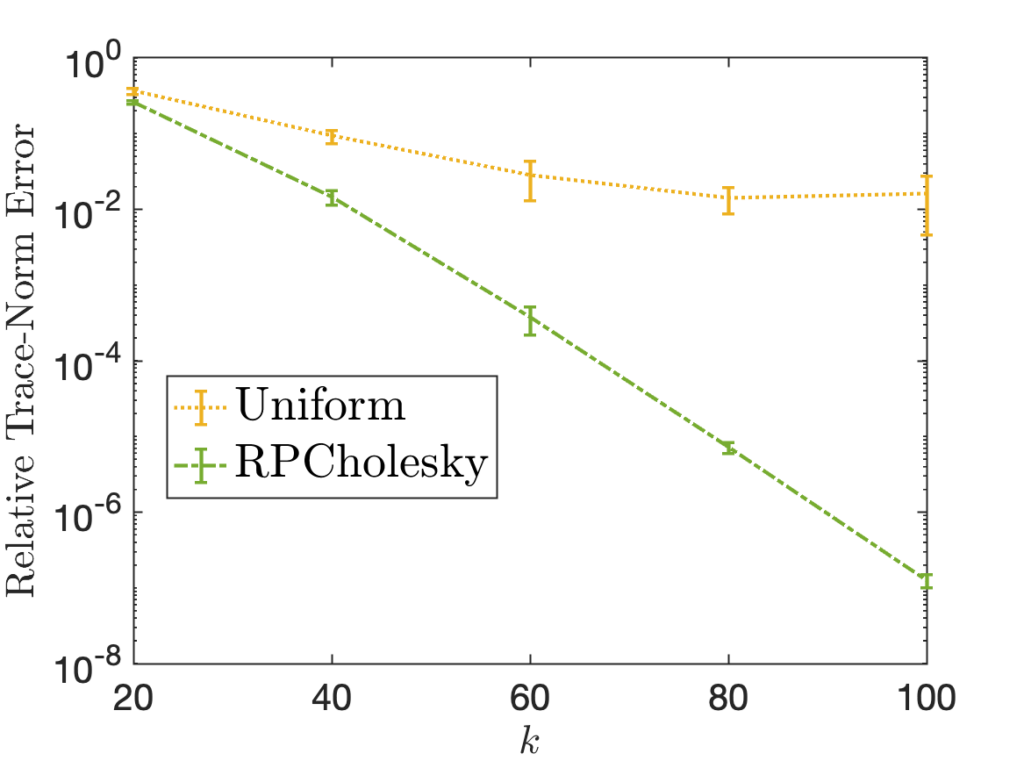
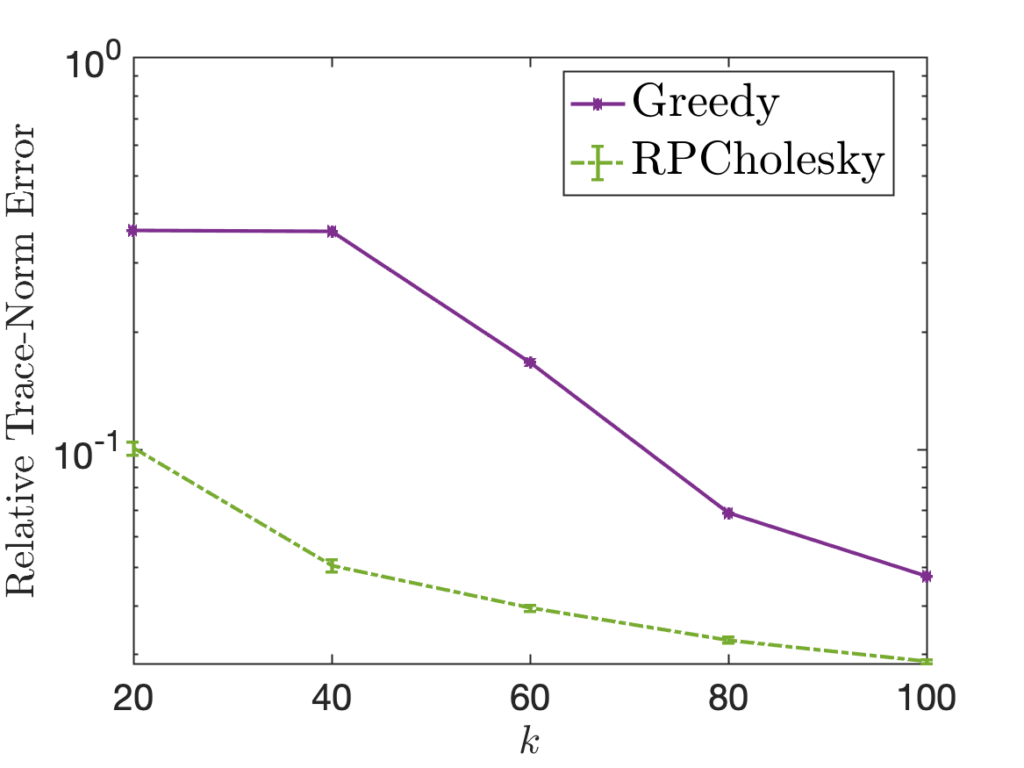
![\[A\langle \Omega\rangle := A\Omega \, (\Omega^*A\Omega)^{-1} \, \Omega^*A. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-636eb074d4ce2e59defede1f1be7877d_l3.png "Rendered by QuickLaTeX.com")
 is invertible; if
is invertible; if  should be replaced by the
should be replaced by the  . For simplicity, I will assume that
. For simplicity, I will assume that  is invertible in this post, though everything we discuss will continue to work if this assumption is dropped. I use
is invertible in this post, though everything we discuss will continue to work if this assumption is dropped. I use  .
. , where
, where  , we observe that the Nyström approximation can be written entirely using
, we observe that the Nyström approximation can be written entirely using ![\[A\langle \Omega\rangle = Y \, (\Omega^* Y)^{-1}\, Y^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4332839cf045146208c9b58c395aefc4_l3.png "Rendered by QuickLaTeX.com")
 is only rank at most
is only rank at most  to be closer to the eigenvectors of
to be closer to the eigenvectors of  of the identity matrix, then
of the identity matrix, then  of
of ![\[A(:,\{i_1,\ldots,i_k\}) = A\Omega \quad \text{for}\quad \Omega = I(:,\{i_1,i_2,\ldots,i_k\}).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49181e8e724ecddcd4646eb90f3bc35e_l3.png "Rendered by QuickLaTeX.com")
 . As a first step, we shall show that the residual is psd. This means that
. As a first step, we shall show that the residual is psd. This means that ![\[A\langle \Omega \rangle = A^{1/2} P_{A^{1/2}\Omega} A^{1/2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4cc583dbaca6e6da9c53ad3327717346_l3.png "Rendered by QuickLaTeX.com")
 denotes the the
denotes the the  . To deduce the projection formula, we break down
. To deduce the projection formula, we break down  in (1):
in (1):![\[A\langle \Omega\rangle = A^{1/2} \left( A^{1/2}\Omega \left[ (A^{1/2}\Omega)^* A^{1/2}\Omega \right]^{-1} (A^{1/2}\Omega)^* \right) A^{1/2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2880bf8a43ee68664131becfdc0b3d19_l3.png "Rendered by QuickLaTeX.com")
 , where
, where  is square and upper triangular. The orthogonal projection is
is square and upper triangular. The orthogonal projection is  . The parenthesized expression is
. The parenthesized expression is  .
.![\[A - A\langle \Omega\rangle = A^{1/2} (I - P_{A^{1/2}\Omega}) A^{1/2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-23f5459646b6b93d24c7e107f636a3d5_l3.png "Rendered by QuickLaTeX.com")
 , if
, if  is psd. If
is psd. If  is an orthogonal projection and therefore psd. Thus, by the conjugation rule, the residual of the is Nyström approximation is psd:
is an orthogonal projection and therefore psd. Thus, by the conjugation rule, the residual of the is Nyström approximation is psd:![\[A - A\langle \Omega\rangle = \left(A^{1/2}\right)^* (I-P_{A^{1/2}\Omega})A^{1/2} \quad \text{is psd}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a06ad324c77c1acb2f4563bff24686a7_l3.png "Rendered by QuickLaTeX.com")
 defined on the set of
defined on the set of ![\[\left\|UBV\right\|_{\rm UI} = \left\|B\right\|_{\rm UI} \quad \text{for all unitary matrices $U$ and $V$.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e3f67a9a35ce0ba62f155eb0cddcf1f2_l3.png "Rendered by QuickLaTeX.com")
 . Unitary matrices preserve the
. Unitary matrices preserve the  . For instance, the spectral, Frobenius, and nuclear norms take the forms
. For instance, the spectral, Frobenius, and nuclear norms take the forms
 , then
, then  for every unitarily invariant norm
for every unitarily invariant norm  , if the difference
, if the difference  is psd. As a consequence,
is psd. As a consequence,  if and only if
if and only if  .
. , it seems natural to expect that
, it seems natural to expect that  for every unitarily invariant norm
for every unitarily invariant norm ![\[\hat{A} = A\Omega \, T \, (A\Omega)^* = A \Omega \, T \, \Omega^*A,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0955f7609913774307be45326f9a1dfd_l3.png "Rendered by QuickLaTeX.com")
 on both sides to obtain
on both sides to obtain![\[\hat{A} = A^{1/2} (A^{1/2}\Omega\, T\, \Omega^*A^{1/2}) A^{1/2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6e53f93f84078b4f7a0c921ee5bb6e8d_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{A} = A^{1/2} \,QMQ^*\, A^{1/2} \quad \text{where} \quad M\text{ is self-adjoint}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-59f6b786fcfd454b3e037d9cfd0a5b50_l3.png "Rendered by QuickLaTeX.com")
![\[A - \hat{A} = A^{1/2} (I - QMQ^*)A^{1/2}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d8a588c5112e24aba21783fd38c417b8_l3.png "Rendered by QuickLaTeX.com")
 is psd (property 3), the conjugation rule tells us that
is psd (property 3), the conjugation rule tells us that![\[I - QMQ^*\succeq 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f5f4a27c026c6cb4a5c756cf9bfea0ac_l3.png "Rendered by QuickLaTeX.com")
 ? We can apply the conjugation rule again to conclude
? We can apply the conjugation rule again to conclude![\[Q^*(I - QMQ^*)Q = Q^*Q - (Q^*Q)M(Q^*Q) = I-M \succeq 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-835a14c1d767ffd164f996bffd8b4de5_l3.png "Rendered by QuickLaTeX.com")
 since
since  . Indeed,
. Indeed,
 and the last line is the conjugation rule combined with the fact that
and the last line is the conjugation rule combined with the fact that  is psd. Thus, having shown
is psd. Thus, having shown![\[A - \hat{A} \succeq A - A\langle\Omega\rangle \succeq 0,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3617754c93689afb140dcd12ad0b7e80_l3.png "Rendered by QuickLaTeX.com")
![\[\|A - \hat{A}\|_{\rm UI} \ge \left\|A - A\langle \Omega\rangle\right\|_{\rm UI} \quad \text{for every unitarily invariant norm $\left\|\cdot\right\|_{\rm UI}$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c89ba0ba57313ce5f778324faf3bff2f_l3.png "Rendered by QuickLaTeX.com")
 where
where  ; our goal in this post will be to derive a fairly general version of the inequality with only explicit constants.
; our goal in this post will be to derive a fairly general version of the inequality with only explicit constants.![\[\mathbb{P}\{|Y|\ge t\} \le \mathrm{e}^{-t^2/a} \quad \text{for some $a>0$ and for all sufficiently large $t$.} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7f713f49e028fa06020270f85a5a1e2f_l3.png "Rendered by QuickLaTeX.com")
 .
. ), then
), then ![\[\xi_Y(t) := \log \mathbb{E} \exp(tY).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c65c7c900f2941726b28371b5fdcb4e8_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_Y(t) \le ca t^2 \quad \text{for all $t\in\mathbb{R}$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e307f36da20e09f721e8f9a24d36a693_l3.png "Rendered by QuickLaTeX.com")
 .
.![\[\xi_{x}(t) \le\frac{1}{2} vt^2 \quad \text{for all $t\in\mathbb{R}$.} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c7c44d1fed75ceb263a286963825c5ae_l3.png "Rendered by QuickLaTeX.com")
 has cgf
has cgf![\[ \xi_X(t) = \frac{1}{2} \sigma^2 t^2, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ee966879f0689027cc0f7f2396dd0dae_l3.png "Rendered by QuickLaTeX.com")
 equal to its variance.
equal to its variance.![\[\mathbb{P}\left\{\left|x^\top Ax - \mathbb{E} \left[x^\top A x\right]\right|\ge t \right\} \le 2\exp\left(- \frac{c\cdot t^2}{v^2\left\|A\right\|_{\rm F}^2 + v\left\|A\right\|t} \right),\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a84a4ab33a1186630c8021cbfcd72d23_l3.png "Rendered by QuickLaTeX.com")
 is a constant (not depending on
is a constant (not depending on  and
and  :
:![\[\mathbb{P}\left\{\left|x^\top Ax - \mathbb{E}\left[x^\top Ax\right]\right|\ge t \right\} \stackrel{\text{small $t$}}{\lessapprox} 2\exp\left(- \frac{c\cdot t^2}{v^2\left\|A\right\|_{\rm F}^2} \right)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-37b39cb580d561ba6777a9e757dc714f_l3.png "Rendered by QuickLaTeX.com")
 :
:![\[\mathbb{P}\left\{\left|x^\top Ax - \mathbb{E}\left[x^\top Ax\right]\right|\ge t \right\} \stackrel{\text{large $t$}}{\lessapprox} 2\exp\left(- \frac{c\cdot t}{v\|A\|} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-244ec27c116074c4ee0033dda85051d8_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{x^\top Ax}(t) \le \frac{16v^2\left\|A\right\|_{\rm F}^2\, t^2}{2(1-4v\left\|A\right\|t)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a024c1e2eb4eb0e9ce3b60a66a5cca04_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ x^\top A x \ge t \} \le \exp\left( -\frac{t^2/2}{16v^2 \left\|A\right\|_{\rm F}^2+4v\left\|A\right\|t} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2213aea3706c09f904897076143402f3_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ x^\top A x \le -t \} \le \exp\left( -\frac{t^2/2}{16v^2 \left\|A\right\|_{\rm F}^2+4v\left\|A\right\|t} \right)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ae296911e4204fa8e70d217568edd442_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ |x^\top A x| \ge t \} \le 2\exp\left( -\frac{t^2/2}{16v^2 \left\|A\right\|_{\rm F}^2+4v\left\|A\right\|t} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4bdfeb16e5c96258871d56cb51203b95_l3.png "Rendered by QuickLaTeX.com")
 at the cost of a factor of four:
at the cost of a factor of four:![\[\xi_{x^\top Ax}(t) \le \xi_{\tilde{x}^\top Ax}(4t). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e20eed7500e7e93630cb1fb1dac324dd_l3.png "Rendered by QuickLaTeX.com")
 to the Gaussian bilinear form
to the Gaussian bilinear form  where
where  and
and  :
:![\[\xi_{g^\top Ag}(t) \le \frac{\left\|A\right\|_{\rm F}^2 \, t^2}{1-2\|A\|\, t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-19f61ae89aa95b37ff49bcbd7ad27b96_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{\tilde{g}^\top Ag}(t) \le \frac{\left\|A\right\|_{\rm F}^2 \, t^2}{2(1-\|A\|\, t)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-38e35e17a1a326c592e4a33717e57bd2_l3.png "Rendered by QuickLaTeX.com")
 to
to  . To do this, we first evaluate the cgf of
. To do this, we first evaluate the cgf of ![\[\log \mathbb{E}_{\tilde{x}} \exp(t \, \tilde{x}^\top Ax) = \sum_{i=1}^n \log \mathbb{E}_{\tilde{x}} \exp(t \,\tilde{x}_i (Ax)_i) \le \frac{1}{2} v \left(\sum_{i=1}^n (Ax)_i^2\right)t^2 \le \frac{1}{2} v\left\|Ax\right\|^2 \, t^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0c463e71b81d7bd984fa8518767f1660_l3.png "Rendered by QuickLaTeX.com")
 in which
in which ![\[\log \mathbb{E}_{\tilde{g}} \exp((\sqrt{v} t) \, \tilde{g}^\top Ax) = \frac{1}{2} v \left\|Ax\right\|^2 \, t^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ef3b5b37870fe8313c9972bbc213f96b_l3.png "Rendered by QuickLaTeX.com")
![\[\log \mathbb{E}_{\tilde{x}} \exp(t \, \tilde{x}^\top Ax) \le \log \mathbb{E}_{\tilde{g}} \exp((\sqrt{v} t) \, \tilde{g}^\top Ax). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c05efe81c8095276378b2e4788d64394_l3.png "Rendered by QuickLaTeX.com")
![\[\log \mathbb{E}_{\tilde{g},x} \exp((\sqrt{v} t) \, \tilde{g}^\top Ax) \le \log \mathbb{E}_{\tilde{g}} \exp \left(\frac{1}{2} v^2 \left\|A^\top \tilde{g}\right\|^2t^2\right) = \log \mathbb{E}_{\tilde{g},g} \exp(v t \, \tilde{g}^\top Ag). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c8fdd18f70e855effd8259473ee86933_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{\tilde{x}^\top Ax}(t)\le \xi_{\tilde{g}^\top Ag}(vt). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fdca14d3f0efedd0b795f0c1f871368c_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{x^\top Ax}(t) \le \xi_{\tilde{x}^\top Ax}(4t) \le \xi_{\tilde{g}^\top Ag}(4vt) \le \frac{16v^2\left\|A\right\|_{\rm F}^2\, t^2}{2(1-4v\left\|A\right\|t)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0037fd7f023f4d3f7058bb77afa263c5_l3.png "Rendered by QuickLaTeX.com")
 for
for  . Then
. Then ![\[\mathbb{P} \left\{ X\ge t \right\} \le \exp\left( -\frac{t^2/2}{v+ct} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-12b977ef8c244a0750402071dcf6caf1_l3.png "Rendered by QuickLaTeX.com")
 to obtain
to obtain![\[\mathbb{P} \{ x^\top A x \le -t \} = \mathbb{P} \{ x^\top (-A) x \ge t \} \le \exp\left( -\frac{t^2/2}{16v^2 \left\|A\right\|_{\rm F}^2+4v\left\|A\right\|t} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f63f2b0759ceb4a78be2f3513ab8ab82_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ |x^\top A x| \ge t \} \le \mathbb{P} \{ x^\top A x \ge t \} + \mathbb{P} \{ x^\top A x \le -t \} \le 2\exp\left( -\frac{t^2/2}{16v^2 \left\|A\right\|_{\rm F}^2+4v\left\|A\right\|t} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f6d5d289322c34b3004c8501cd9cf477_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{x^\top Ax-\mathbb{E} [x^\top A x]}(t) \le \frac{40v^2\left\|A\right\|_{\rm F}^2\, t^2}{2(1-8v\left\|A\right\|t)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9ec7d119e890e1feaf86137b543c2adc_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ x^\top A x-\mathbb{E} [x^\top A x] \ge t \} \le \exp\left( -\frac{t^2/2}{40v^2 \left\|A\right\|_{\rm F}^2+8v\left\|A\right\|t} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5d35dd74f9d307d1616a4c0b2d10a7ac_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ x^\top A x-\mathbb{E} [x^\top A x] \le -t \} \le \exp\left( -\frac{t^2/2}{40v^2 \left\|A\right\|_{\rm F}^2+8v\left\|A\right\|t} \right)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4d0c27970de892090fbb22751582e8a0_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{P} \{ |x^\top A x-\mathbb{E} [x^\top A x]| \ge t \} \le 2\exp\left( -\frac{t^2/2}{40v^2 \left\|A\right\|_{\rm F}^2+8v\left\|A\right\|t} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0239d1a1656250c1cdb5b0c5557973e4_l3.png "Rendered by QuickLaTeX.com")
 into its diagonal and off-diagonal portions. For any two random variables
into its diagonal and off-diagonal portions. For any two random variables ![\begin{align*} \xi_{X+Y}(t) &= \log \mathbb{E} \left[\exp(tX)\exp(tY)\right] \\&\le \log \left(\left[\mathbb{E} \exp(2tX)\right]^{1/2}\left[\mathbb{E}\exp(2tY)\right]^{1/2}\right) \\&=\frac{1}{2} \xi_X(2t) + \frac{1}{2}\xi_Y(2t). \end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5fd6293d750d7bd98a31189e5802f59e_l3.png "Rendered by QuickLaTeX.com")
![x^\top D x - \mathbb{E}[x^\top Ax]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c7bc48f04ae3847d7b03462151fca3f7_l3.png "Rendered by QuickLaTeX.com") and
and  . We begin with the diagonal part. We compute
. We begin with the diagonal part. We compute![\begin{align*}\xi_{x^\top D x - \mathbb{E}[x^\top Ax]}(t) &= \log \mathbb{E} \exp\left(t \sum_{i=1}^n A_{ii}(x_i^2 - \mathbb{E}[x_i^2]) \right) \\ &= \sum_{i=1}^n \log \mathbb{E} \exp\left((t A_{ii})\cdot(x_i^2 - \mathbb{E}[x_i^2]) \right). \end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-03a1f2a90412fa1e00f7deb32d479def_l3.png "Rendered by QuickLaTeX.com")
![x_i^2 - \mathbb{E}[x_i^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2ceca9ceddd7db5d803529c2da5c349c_l3.png "Rendered by QuickLaTeX.com") , we use the following bound, taken from
, we use the following bound, taken from ![\[\log \mathbb{E} \exp\left(t(x_i^2 - \mathbb{E}[x_i^2]) \right) \le \frac{8v^2t^2}{1-2v|t|}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec7de84a4e1f848cf06fb619582b4418_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{x^\top D x - \mathbb{E}[x^\top Ax]}(t) \le \sum_{i=1}^n \frac{8v^2|A_{ii}|^2t^2}{1-2v|A_{ii}|t} \le \frac{8v^2\|A\|_{\rm F}^2t^2}{1-2v\|A\|t}\quad \text{for $t>0$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-50a71b505cc381540a6934a28416f614_l3.png "Rendered by QuickLaTeX.com")
 and
and  .
. , where we’ve both replaced one copy of
, where we’ve both replaced one copy of ![\[\xi_{x^\top F x}(t) \le \xi_{\tilde{x}^\top Ax}(4t).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a6c678fd5b04c01bb1001d4f6897493_l3.png "Rendered by QuickLaTeX.com")
![\[ \xi_{x^\top Fx}(t) \le \frac{16v^2\left\|A\right\|_{\rm F}^2\, t^2}{2(1-4v\left\|A\right\|t)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-29b5b5a2aa3fe27580abf965005db1d0_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\xi_{x^\top Ax-\mathbb{E} [x^\top A x]} &\le \frac{1}{2} \xi_{x^\top D x - \mathbb{E}[x^\top Ax]}(2t) + \frac{1}{2} \xi_{x^\top Fx}(2t) \\&\le \frac{8v^2\|A\|_{\rm F}^2t^2}{2(1-4v\|A\|t)} + \frac{32v^2\left\|A\right\|_{\rm F}^2\, t^2}{2(1-8v\left|A\right|t)} \\&\le \frac{8v^2\|A\|_{\rm F}^2t^2}{2(1-4v\|A\|t)} + \frac{32v^2\left\|A\right\|_{\rm F}^2\, t^2}{2(1-8v\left\|A\right\|t)} \\&\le \frac{40v^2\left\|A\right\|_{\rm F}^2\, t^2}{2(1-8v\left\|A\right\|t)}.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e26ce05b781b47ce078f0addb9559240_l3.png "Rendered by QuickLaTeX.com")
 :
:
 .
. :
:
 on the interval
on the interval ![[-\pi,\pi]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-db4571552bede65e472ffbfee6887855_l3.png "Rendered by QuickLaTeX.com") to visually near-perfect accuracy by connecting the dots between seven points
to visually near-perfect accuracy by connecting the dots between seven points  :
:
 on the interval
on the interval ![[-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-634fe4705a526f221e312cffd3ceda93_l3.png "Rendered by QuickLaTeX.com") :
: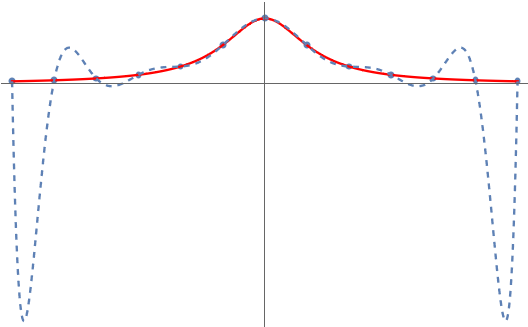
 is terrible! What’s going on? Why doesn’t polynomial interpolation work here? Can we fix it? The answer to the last question is yes and the solution is Chebyshev polynomials.
is terrible! What’s going on? Why doesn’t polynomial interpolation work here? Can we fix it? The answer to the last question is yes and the solution is Chebyshev polynomials.

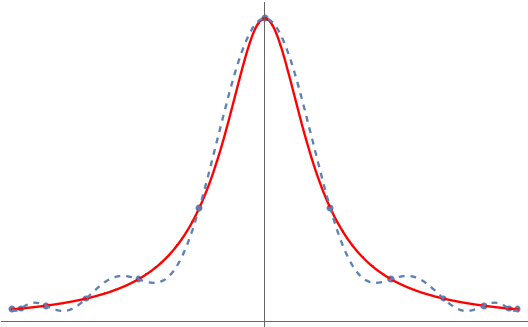
 , like this for ourselves. We’ll assume the interpolation points are given by a function
, like this for ourselves. We’ll assume the interpolation points are given by a function  so we can form the polynomial interpolant for any desired polynomial degree
so we can form the polynomial interpolant for any desired polynomial degree  being \textit{injective} so that the points
being \textit{injective} so that the points  are guaranteed to be distinct.
are guaranteed to be distinct. , the points look like this:
, the points look like this:

 (shown on horizontal axis)
(shown on horizontal axis) .
.
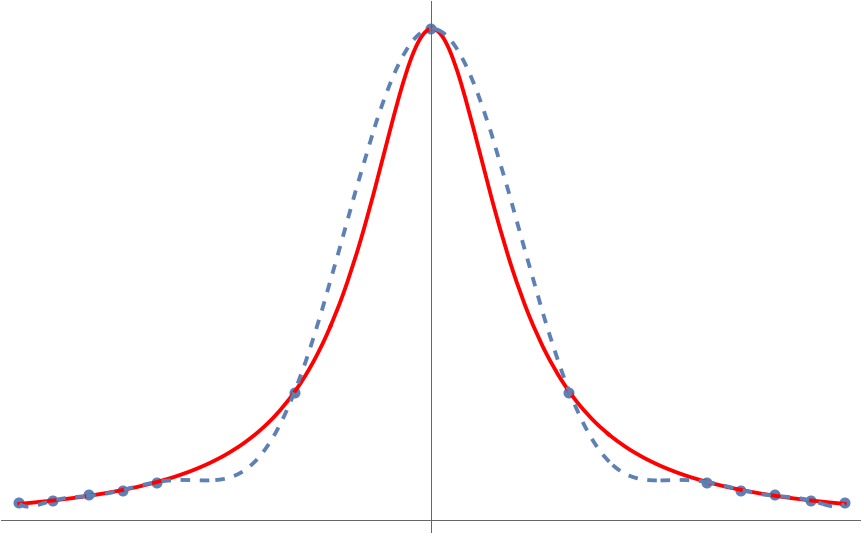
 , we see that these interpolation points now lead to a failure to approximate the function at the middle of the interval, even if we use a lot of interpolation points!
, we see that these interpolation points now lead to a failure to approximate the function at the middle of the interval, even if we use a lot of interpolation points!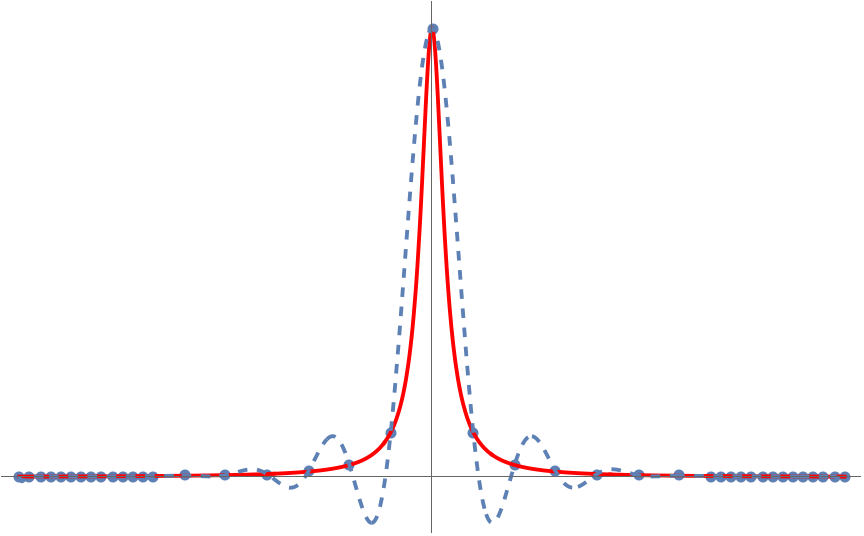
 and
and  are, the more clustered the points will be. For instance,
are, the more clustered the points will be. For instance,


 , and
, and ,
,  .
. . This function yields points which are more clustered at the endpoints:
. This function yields points which are more clustered at the endpoints:


 which not too rough (i.e.,
which not too rough (i.e.,  gives the
gives the  radians along the unit circle. This means that the Chebyshev points are the
radians along the unit circle. This means that the Chebyshev points are the  ).
).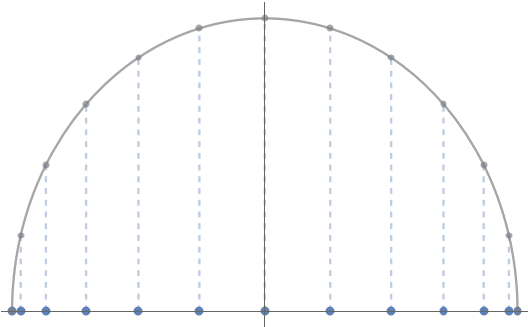
 look like as a function of the angle
look like as a function of the angle  to
to 
 , is a polynomial in the variable
, is a polynomial in the variable 
 always take the form
always take the form

 units of frequency
units of frequency  units of frequency
units of frequency  units of frequency
units of frequency  with exponentially small errors in the number of interpolation points used. By contrast, functions with kinks like
with exponentially small errors in the number of interpolation points used. By contrast, functions with kinks like  are approximated with errors which decay much more slowly. See
are approximated with errors which decay much more slowly. See 
 is
is  since multiple
since multiple  , between which the cosine function is
, between which the cosine function is 
 , we’ve instead written
, we’ve instead written 
 are known as the
are known as the  are known as the Chebyshev polynomials
are known as the Chebyshev polynomials 

 for
for 
 and
and  are both polynomials, every Chebyshev polynomial is as well.
are both polynomials, every Chebyshev polynomial is as well.![\[p^\circ(\theta) = b_n\cos(n\theta) + \cdots + b_1\cos\theta + b_0\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c6828300bbae5fa0051e7c134da1d61_l3.png "Rendered by QuickLaTeX.com")
![\[p(x) = b_nT_n(x) + \cdots + b_1 T_1(x) + b_0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cb291d9f73ebc4e74d0455dac4558a48_l3.png "Rendered by QuickLaTeX.com")
 are orthogonal to each other; are the Chebyshev polynomials?
are orthogonal to each other; are the Chebyshev polynomials?
 triplets and checking whether they form a triangle is far too computationally costly for the billions of Facebook accounts. Somehow, we want to do much faster than this, and to achieve this speed we would be willing to settle for an estimate of the triangle count up to some error.
triplets and checking whether they form a triangle is far too computationally costly for the billions of Facebook accounts. Somehow, we want to do much faster than this, and to achieve this speed we would be willing to settle for an estimate of the triangle count up to some error. matrix where the
matrix where the  th entry of
th entry of  of the matrix
of the matrix  where
where  , and
, and  , which is twice the number of triangles incident on
, which is twice the number of triangles incident on  and
and  are both counted as paths of length 3 for a triangle consisting of
are both counted as paths of length 3 for a triangle consisting of  is counted thrice in the
is counted thrice in the  th, and
th, and  th entries of
th entries of 
 for a vector
for a vector  . Therefore, the matrix
. Therefore, the matrix  , we simply compute matrix–vector products with
, we simply compute matrix–vector products with  .
. and
and  occurs with equal
occurs with equal  . Since the entries of
. Since the entries of  and
and  are independent, the
are independent, the  is
is  is
is![\begin{equation*} \mathbb{E} \, x^\top M x = \sum_{i,j=1}^n M_{ij} \mathbb{E} [x_ix_j] = \sum_{i = 1}^n M_{ii} = \operatorname{tr}(M). \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-17a6a7cacc94a45f13853dc8f34f1e3a_l3.png "Rendered by QuickLaTeX.com")
 . Thus, the efficiently computable quantity
. Thus, the efficiently computable quantity  equals
equals  and compute the averaged trace estimator
and compute the averaged trace estimator
 remains an unbiased estimator for
remains an unbiased estimator for  , but with reduced variability. Quantitatively, the
, but with reduced variability. Quantitatively, the 
 .” In other words, concentration inequalities can provide quantitative estimates of the likely size of the error when a randomized algorithm is executed.
.” In other words, concentration inequalities can provide quantitative estimates of the likely size of the error when a randomized algorithm is executed. is bounded by the expected value of
is bounded by the expected value of 
 . This small example shows both the power and the limitation of Markov’s inequality. On the negative side, our analysis suggests that we might have to wait as much as 100 times the average runtime for the algorithm to complete running with 99% probability; this large huge multiple of 100 seems quite pessimistic. On the other hand, we needed no information whatsoever about how the algorithm works to do this analysis. In general, Markov’s inequality cannot be improved without more assumptions on the random variable
. This small example shows both the power and the limitation of Markov’s inequality. On the negative side, our analysis suggests that we might have to wait as much as 100 times the average runtime for the algorithm to complete running with 99% probability; this large huge multiple of 100 seems quite pessimistic. On the other hand, we needed no information whatsoever about how the algorithm works to do this analysis. In general, Markov’s inequality cannot be improved without more assumptions on the random variable  :
:
 denote the average
denote the average

 . Therefore, by Chebyshev’s inequality,
. Therefore, by Chebyshev’s inequality,
 and are willing to tolerate a failure probability of
and are willing to tolerate a failure probability of 
 . Normal random variables have an exponentially small probability of being more than a few standard deviations above their mean, so it is natural to expect this should be true of
. Normal random variables have an exponentially small probability of being more than a few standard deviations above their mean, so it is natural to expect this should be true of 
 . Hoeffding’s inequality makes the assumption that the summands are bounded, say within an interval
. Hoeffding’s inequality makes the assumption that the summands are bounded, say within an interval ![[a,b]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-336bb22d4b2092329486008f6665b888_l3.png "Rendered by QuickLaTeX.com") .
.
 replaced by the potentially much larger quantity
replaced by the potentially much larger quantity .
. for every
for every  . We continue to denote
. We continue to denote 

 . We conclude that Bernstein’s inequality provides sharper bounds then Hoeffding’s inequality for smaller values of
. We conclude that Bernstein’s inequality provides sharper bounds then Hoeffding’s inequality for smaller values of  , except with failure probability at most
, except with failure probability at most 

![[\mu-B,\mu+B]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bada0c6b976891e43bcc4c3b7508e96e_l3.png "Rendered by QuickLaTeX.com") for every
for every  , then
, then
 samples are needed rather than proportional to
samples are needed rather than proportional to  . The fact that we need proportional to
. The fact that we need proportional to  samples to achieve error
samples to achieve error  to
to  , which is a huge improvement.
, which is a huge improvement. ; for small values of
; for small values of 
 is the
is the 
 . By the
. By the  and
and  where
where  and
and  are the
are the  is the
is the  so
so  and
and  . Since the
. Since the 


 and
and  . Since we often don’t know good bounds for
. Since we often don’t know good bounds for  , one should really use the trace estimator together with an
, one should really use the trace estimator together with an  down from
down from  .
. is an
is an 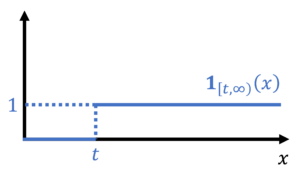
![\begin{equation*} \mathbb{P}\{X \ge t \} = \mathbb{E}[\mathbf{1}_{[t,\infty)}(X)]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-070609e7e86c3846db2eec810355d5f4_l3.png "Rendered by QuickLaTeX.com")
 by bounding its corresponding indicator function. In particular, we have the inequality
by bounding its corresponding indicator function. In particular, we have the inequality 
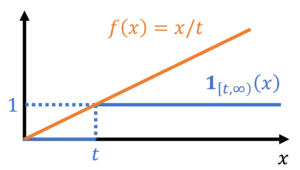
![\begin{equation*} \mathbb{P}\{ X \ge t \} = \mathbb{E}[\mathbf{1}_{[t,\infty)}(X)] \le \mathbb{E} \left[ \frac{X}{t} \right] = \frac{\mathbb{E} X}{t}. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49a753eda4e591b364f5d4f3e24a0b9b_l3.png "Rendered by QuickLaTeX.com")

 by (11). To obtain sharp and useful bounds on
by (11). To obtain sharp and useful bounds on  we seek bounding functions
we seek bounding functions  ,
,  should be close to zero,
should be close to zero, ,
,  to be easily computable or boundable.
to be easily computable or boundable. or exponentials
or exponentials  for which we have hopes of computing or bounding
for which we have hopes of computing or bounding  (by increasing
(by increasing  , detracting from our ability to achieve point 2. We shall eventually come up with a best-possible resolution to this dilemma by formulating this as an optimization problem to determine the best choice of the parameter
, detracting from our ability to achieve point 2. We shall eventually come up with a best-possible resolution to this dilemma by formulating this as an optimization problem to determine the best choice of the parameter  to obtain the best possible candidate function of the given form.
to obtain the best possible candidate function of the given form. satisfies the bound (13), and thus by (12),
satisfies the bound (13), and thus by (12),
 , is a nonnegative random variable. Thus applying (14) gives
, is a nonnegative random variable. Thus applying (14) gives
 if, and only if,
if, and only if,  . Therefore, by Markov’s inequality,
. Therefore, by Markov’s inequality,
 , where
, where 

 ,
,  , and the
, and the 
 up to the sign of the parameter
up to the sign of the parameter  . Taking logarithms proves the additivity.
. Taking logarithms proves the additivity.
 and consider the cumulant generating function of
and consider the cumulant generating function of  . Then one can show the cumulant generating function bound
. Then one can show the cumulant generating function bound . We have
. We have  . Since the function
. Since the function  is
is  . Taking expectations, we have
. Taking expectations, we have  . One can show by comparing Taylor series that
. One can show by comparing Taylor series that  . Therefore, we have
. Therefore, we have  .
.



 :
:
 , we can apply a small trick. If we apply (20) to the summands
, we can apply a small trick. If we apply (20) to the summands  instead of
instead of 
 . The means of doing often this goes by the fancy name
. The means of doing often this goes by the fancy name 


 .
.