I am delighted to share that my paper Randomly pivoted Cholesky: Randomly pivoted Cholesky: Practical approximation of a kernel matrix with few entry evaluations, joint with Yifan Chen, Joel Tropp, and Robert Webber, has been published online in Communications on Pure and Applied Mathematics. To celebrate this occasion, I want to share one of my favorite tricks in the design of low-rank approximation algorithms, which I will call the Gram correspondence.
Projection Approximation and Nyström Approximation
When we construct a low-rank approximation to a matrix, the type of approximation we use is typically dictated by the size and properties of the matrix. For a rectangular matrix  , one of the standard techniques is the standard randomized SVD algorithm:
, one of the standard techniques is the standard randomized SVD algorithm:
- Generate a Gaussian random matrix
 .
.
- Form the product
 .
.
- Compute an (economy-size) QR decomposition
 .
.
- Evaluate the SVD
 .
.
- Output the low-rank approximation
 for
for  .
.
Succinctly, the output of the randomized SVD is given by the formula  , where
, where  denotes the orthogonal projector onto the column span of a matrix
denotes the orthogonal projector onto the column span of a matrix  . This motivates the general definition:
. This motivates the general definition:
Definition (projection approximation): Given a test matrix , the projection approximation to the matrix  is .
is .
The class of projection approximations is much richer than merely the approximation constructed by the standard randomized SVD. Indeed, low-rank approximations computed by randomized subspace iteration, randomized block Krylov iteration, column-pivoted QR decompositions, etc. all fit under the umbrella of projection approximations.
Many matrices in applications have additional structure such as symmetry or sparsity, and it can be valuable to make use of low-rank approximations that take advantage of those properties. An especially important type of structure is positive semidefiniteness. For our purposes, a positive semidefinite matrix  is one that is symmetric and possesses nonnegative eigenvalues, and we will use abbreviate “positive semidefinite” as “psd”. Psd matrices arise in applications as covariance matrices, kernel matrices, and as discretizations of certain differential and integral operators. Further, any rectangular matrix gives rise to its psd Gram matrix
is one that is symmetric and possesses nonnegative eigenvalues, and we will use abbreviate “positive semidefinite” as “psd”. Psd matrices arise in applications as covariance matrices, kernel matrices, and as discretizations of certain differential and integral operators. Further, any rectangular matrix gives rise to its psd Gram matrix  ; we will have much more to say about Gram matrices below.
; we will have much more to say about Gram matrices below.
To compute a low-rank approximation to a psd matrix, the preferred format as usually the Nyström approximation:
Definition (Nyström approximation): Given a test matrix , the Nyström approximation is
![\[\hat{A} \coloneqq (A\Omega) (\Omega^\top A \Omega)^{-1}(A\Omega)^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-345eb2a18a6428fbbe9cfa2d305a3026_l3.png "Rendered by QuickLaTeX.com")
As discussed in a previous post, the general class of Nyström approximations includes many useful specializations depending on how the matrix  is selected.
is selected.
The Gram Correspondence
The Gram correspondence is a connection between projection approximation and Nyström approximation:
The Gram correspondence: Let  be any rectangular matrix and consider the Gram matrix . Fix a test matrix , and define the Nyström approximation
be any rectangular matrix and consider the Gram matrix . Fix a test matrix , and define the Nyström approximation  to
to  and the projection approximation
and the projection approximation  to . Then
to . Then
![\[\hat{A} = \smash{\hat{B}}^\top \hat{B}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-70de4cbe941b96a1dc7655f240027afd_l3.png "Rendered by QuickLaTeX.com")
That is, the Gram matrix  of the projection approximation
of the projection approximation  is the Nyström approximation
is the Nyström approximation  .
.
As we will see, this correspondence has many implications:
- Special cases. The correspondence contains several important facts as special cases. Examples include the equivalence between the randomized SVD and single-pass Nyström approximation and the equivalence of (partial) column-pivoted QR factorization and (partial) pivoted Cholesky decomposition.
- Algorithm design. Since Nyström approximation and projection approximations are closely related, one can often interconvert algorithms for one type of approximation into the other. These conversions can lead one to discover new algorithms. We will provide a historical answer with the discovery of randomly pivoted Cholesky.
- Error bounds. The Gram correspondence is immensely helpful in the analysis of algorithms, as it makes error bounds for projection approximations and Nyström approximations easily derivable from each other.
For those interested, we give a short proof of the Gram correspondence below.
Proof of Gram correspondence
The Gram correspondence is straightforward to derive, so let’s do so before moving on. Because

is a projection matrix, it satisfies

. Thus,
![\[\hat{B}^\top \hat{B} = B^\top \Pi_{B\Omega}^2B = B^\top \Pi_{B\Omega}B.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fd1a575e449aa0954d4f62e109d824fe_l3.png "Rendered by QuickLaTeX.com")
The projector
has the formula

. Using this formula, we obtain
![\[\hat{B}^\top \hat{B} = B^\top B\Omega (\Omega^\top B^\top B \Omega)^{-1} \Omega^\top B^\top B.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f3a9aa2fc08a78569f1745222d2565d5_l3.png "Rendered by QuickLaTeX.com")
This is precisely the formula for the Nyström approximation

, confirming the Gram correspondence.
Aside: Gram Square Roots
Before we go forward, let me highlight a point of potential conclusion and introduce some helpful terminology. When thinking about algorithms for a psd matrix , it will often to be helpful to conjure a matrix for which . Given a psd matrix , there is always a matrix for which , but this is not unique. Indeed, if , then we have the infinite family of decompositions  generated by every matrix
generated by every matrix  with orthonormal columns. This motivates the following definition:
with orthonormal columns. This motivates the following definition:
Definition (Gram square root): A matrix is a Gram square root for if .
A Gram square root for need not be a square root in the traditional sense that  . Indeed, a Gram square root can be rectangular, so
. Indeed, a Gram square root can be rectangular, so  need not even be defined. Using the Gram square root terminology, the Gram correspondence can be written more succinctly:
need not even be defined. Using the Gram square root terminology, the Gram correspondence can be written more succinctly:
Gram correspondence (concise): If is any Gram square root of , then the projection approximation is a Gram square root of the Nyström approximation  .
.
Examples of the Gram Correspondence
Before going further, let us see a couple of explicit examples of the Gram correspondence.
Randomized SVD and Single-Pass Nyström Approximation
The canonical example of the Gram correspondence is the equivalence of the randomized SVD algorithm and the single-pass Nyström approximation. Let be a Gram square root for a psd matrix . The randomized SVD of is given by the following steps, which we saw in the introduction:
- Generate a Gaussian random matrix .
- Form the product .
- Compute a QR decomposition .
- Evaluate the SVD .
- Output the low-rank approximation for .
Now, imagine taking the same Gaussian matrix from the randomized SVD, and use it to compute the Nyström approximation . Notice that this Nyström approximation can be computed in a single pass over the matrix . Namely, use a single pass over to compute  and use following formula:
and use following formula:
![\[\hat{A} = Y (\Omega^\top Y)^{-1} Y^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-439e3ea265c77f2fcf84c6b310f185aa_l3.png "Rendered by QuickLaTeX.com")
For this reason, we call

the single-pass Nyström approximation.
The Gram correspondence tells us that the randomized SVD and single-pass Nyström approximation are closely related, in the sense that  . The randomized SVD approximation
. The randomized SVD approximation  is a Gram square root of the single-pass Nyström approximation .
is a Gram square root of the single-pass Nyström approximation .
Column-Pivoted QR Factorization and Pivoted Cholesky Decomposition
A more surprising consequence of the Gram correspondence is the connection between low-rank approximations produced by partial column-pivoted QR factorization of a rectangular matrix and a partial pivoted Cholesky decomposition of . Let’s begin by describing these two approximation methods.
Let’s begin with pivoted partial Cholesky decomposition. Let be a psd matrix, and initialize the zero approximation  . For
. For  , perform the following steps:
, perform the following steps:
- Choose a column index
 . These indices are referred to as pivot indices or, more simply, pivots.
. These indices are referred to as pivot indices or, more simply, pivots.
- Update the low-rank approximation
![\[\hat{A} \gets \hat{A} + \frac{A(:,s_i)A(s_i,:)}{A(s_i,s_i)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-863d08314865254415f4bb93e143faa9_l3.png "Rendered by QuickLaTeX.com")
- Update the matrix
![\[A \gets A - \frac{A(:,s_i)A(s_i,:)}{A(s_i,s_i)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9c0c04b9e4c6217f257a30771739903c_l3.png "Rendered by QuickLaTeX.com")
Here, we are using MATLAB notation to index the matrix . The output of this procedure is
![\[\hat{A} = A(:,S) A(S,S)^{-1} A(S,:),\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bbbddde5089193acc3707f2cc631eddd_l3.png "Rendered by QuickLaTeX.com")
where

. This type of low-rank approximation is known as a
column Nyström approximation and is a special case of the general Nyström approximation with test matrix

equal to a subset of columns of the identity matrix

. For an explanation of why this procedure is called a “pivoted partial Cholesky decomposition” and the relation to the usual notion of Cholesky decomposition, see
this previous post of mine.
Given the Gram correspondence, we expect that this pivoted partial Cholesky procedure for computing a Nyström approximation to a psd matrix should have an analog for computing a projection approximation to a rectangular matrix . This analog is given by the partial column-pivoted QR factorization, which produces a low-rank approximation according as follows. Let be a psd matrix, and initialize the zero approximation  . For , perform the following steps:
. For , perform the following steps:
- Choose a pivot index .
- Update the low-rank approximation by adding the projection of onto the selected column:
 .
.
- Update the matrix by removing the projection of onto the selected column:
 .
.
The output of this procedure is the column projection approximation  , which is an example of the general projection approximation with .
, which is an example of the general projection approximation with .
The pivoted partial Cholesky and QR factorizations are very traditional ways of computing a low-rank approximation in numerical linear algebra. The Gram correspondence tells us immediately that these two approaches are closely related:
Let be a Gram square root of . Compute a column projection approximation to using a partially column-pivoted QR factorization with pivots  , and compute a column Nyström approximation to using a partial pivoted Cholesky decomposition with the same set of pivots . Then
, and compute a column Nyström approximation to using a partial pivoted Cholesky decomposition with the same set of pivots . Then  .
.
I find this remarkable: the equivalence of the randomized SVD and single-pass randomized Nyström approximation and the equivalence of (partial pivoted) Cholesky and QR factorizations are both consequences of the same general principle, the Gram correspondence.
Using the Gram Correspondence to Discover Algorithms
The Gram correspondence is more than just an interesting way of connecting existing types of low-rank approximations: It can be used to discover new algorithms. We can illustrate this with an example, the randomly pivoted Cholesky algorithm.
Background: The Randomly Pivoted QR Algorithm
In 2006, Deshpande, Rademacher, Vempala, and Wang discovered an algorithm that they called adaptive sampling for computing a projection approximation to a rectangular matrix . Beginning from the trivial initial approximation , algorithm proceeds as follows for :
- Choose a column index randomly according to the squared column norm distribution:
![\[\prob\{s_i = j\} = \frac{\norm{B(:,j)}^2}{\norm{B}_{\rm F}^2} \quad \text{for } j=1,2,\ldots,k.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-69b8b3115bb28983cf8ffde1e706bbad_l3.png "Rendered by QuickLaTeX.com")
- Update the low-rank approximation by adding the projection of onto the selected column: .
- Update the matrix by removing the projection of onto the selected column: .
Given our discussion above, we recognize this as an example of partial column-pivoted QR factorization with a particular randomized procedure for selecting the pivots  . Therefore, it can also be convenient to call this algorithm randomly pivoted QR.
. Therefore, it can also be convenient to call this algorithm randomly pivoted QR.
Randomly pivoted QR is a nice algorithm for rectangular low-rank approximation. Each step requires a full pass over the matrix and  operations, so the full procedure requires
operations, so the full procedure requires  operations. This makes the cost of the algorithm similar to other methods for rectangular low-rank approximation such as the randomized SVD, but it has the advantage that it computes a column projection approximation. However, randomly pivoted QR is not a particularly effective algorithm for computing a low-rank approximation to a psd matrix, since—as we shall see—there are faster procedures available.
operations. This makes the cost of the algorithm similar to other methods for rectangular low-rank approximation such as the randomized SVD, but it has the advantage that it computes a column projection approximation. However, randomly pivoted QR is not a particularly effective algorithm for computing a low-rank approximation to a psd matrix, since—as we shall see—there are faster procedures available.
Following the Gram correspondence, we expect there should be an analog of the randomly pivoted QR algorithm for computing a low-rank approximation of a psd matrix. That algorithm, which we call randomly pivoted Cholesky, is derived from randomly pivoted QR in the following optional section:
Derivation of Randomly Pivoted Cholesky Algorithm
Let
be a psd matrix. For conceptual purposes, let us also consider a Gram square root

of
; this matrix
will only be a conceptual device that we will use to help us derive the appropriate algorithm, not something that will be required to run the algorithm itself. Let us now walk through the steps of the randomly pivoted QR algorithm on
, and see how they lead to a
randomly pivoted Cholesky algorithm for computing a low-rank approximation to
.
Step 1 of randomly pivoted QR. Randomly pivoted QR begins by drawing a random pivot according to the rule
Since
, we may compute
![\[\norm{B(:,j)}^2 = B(:,j)^\top B(:,j) = (B^\top B)(j,j) = A(j,j).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8cc2adf4cfa7b83d8d6cf2f459be5e18_l3.png "Rendered by QuickLaTeX.com")
The squared column norms of
are the diagonal entries of the matrix
. Similarly,

. Therefore, we can write the probability distribution for the random pivot
using only the matrix
as
![\[\prob\{s_i = j\} = \frac{A(j,j)}{\tr A} \quad \text{for } j=1,2,\ldots,N.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-203e590f41ae6a725f52f179d8c00c7c_l3.png "Rendered by QuickLaTeX.com")
Step 2 of randomly pivoted QR. The randomly pivoted QR update rule is . Therefore, the update rule for  is
is
![\[\hat{A} &\gets \left(\hat{B} + \frac{B(:,s_i) (B(:,s_i)^\top B)}{\norm{B(:,s_i)}^2}\right)^\top \left(\hat{B} + \frac{B(:,s_i) (B(:,s_i)^\top B)}{\norm{B(:,s_i)}^2}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c91dc552516b91b718159b2a4a798ed3_l3.png "Rendered by QuickLaTeX.com")
After a short derivation,, this simplifies to the update rule
![\[\hat{A} \gets \hat{A} +\frac{A(:,s_i)A(s_i,:)}{A(s_i,s_i)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f5b4e7c853586b6bd3ae380ec4c9f9fb_l3.png "Rendered by QuickLaTeX.com")
Two remarkable things have happened. First, we have obtained an update rule for
that depends only on the psd matrices
and
, and all occurences of the Gram square roots
and
have vanished. Second, this update rule is exactly the update rule for a partial Cholesky decomposition.
Step 3 of randomly pivoted QR. Using a similar derivation to step 2, we update an update rule for :
![\[A \gets A -\frac{A(:,s_i)A(s_i,:)}{A(s_i,s_i)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c5812e0143a84e8e070ef048c409df97_l3.png "Rendered by QuickLaTeX.com")
This completes the derivation.
Randomly Pivoted Cholesky
That derivation was a bit complicated, so let’s summarize. We can with the randomly pivoted QR algorithm for computing a projection approximation to a rectangular matrix , and we used it to derive a randomly pivoted Cholesky algorithm for computing a column Nyström approximation to a psd matrix . Removing the cruft of the derivation, this algorithm is very simple to state:
- Choose a column index randomly according to the diagonal distribution:
![\[\prob\{s_i = j\} = \frac{A(j,j)}{\tr(A)} \quad \text{for } j=1,2,\ldots,k.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-284bb185bf576727c1adf2287ce110e7_l3.png "Rendered by QuickLaTeX.com")
- Update the low-rank approximation
- Update the matrix
I find this to be remarkably cool. We started with a neat algorithm (randomly pivoted QR) for approximating rectangular matrices, and we used the Gram correspondence to derive a different randomly pivoted Cholesky algorithm for psd matrix approximation!
And randomly pivoted Cholesky allows us to pull a cool trick that we couldn’t with randomly pivoted QR. Observe that the randomly pivoted Cholesky algorithm only ever interacts with the residual matrix through the selected pivot columns  and through the diagonal entries
and through the diagonal entries  . Therefore, we can derive an optimized version of the randomly pivoted Cholesky algorithm that only reads
. Therefore, we can derive an optimized version of the randomly pivoted Cholesky algorithm that only reads  entries of the matrix (
entries of the matrix ( columns plus the diagonal) and requires only
columns plus the diagonal) and requires only  operations! See our paper for details.
operations! See our paper for details.
So we started with the randomly pivoted QR algorithm, which requires  operations, and we used it to derive the randomly pivoted Cholesky algorithm that runs in
operations, and we used it to derive the randomly pivoted Cholesky algorithm that runs in  operations. Let’s make this concrete with some specific numbers. Setting
operations. Let’s make this concrete with some specific numbers. Setting  and
and  , randomly pivoted QR requires roughly
, randomly pivoted QR requires roughly  (100 trillion) operations and randomly pivoted Cholesky requires roughly
(100 trillion) operations and randomly pivoted Cholesky requires roughly  (10 billion) operations, a factor of 10,000 smaller operation count!
(10 billion) operations, a factor of 10,000 smaller operation count!
Randomly pivoted Cholesky has an interesting history. It appears to have first appeared in print in the work of Musco and Woodruff (2017), who used the Gram correspondence to derive the algorithm from randomly pivoted QR. It is remarkable that it took a full 11 years after Deshpande and co-author’s original work on randomly pivoted QR for randomly pivoted Cholesky to be discovered. Even after Musco and Woodruff’s paper, the algorithm appears to have largely been overlooked for computation in practice, and I am unaware of any paper documenting computational experiments with randomly pivoted Cholesky before our paper was initially released in 2022. Our paper reexamined the randomly pivoted Cholesky procedure, providing computational experiments comparing it to alternatives and using it for scientific machine learning applications, and provided new error bounds.
Other Examples of Algorithm Design by the Gram Correspondence
The Gram correspondence gives a powerful tool for inventing new algorithms or showing equivalence between existing algorithms. There are many additional examples, such as block (and rejection sampling-accelerated) versions of randomly pivoted QR/Cholesky, Nyström versions of randomized block Krylov iteration, and column projection/Nyström approximations generated by determinantal point process sampling. Indeed, if you invent a new low-rank approximation algorithm, it is always worth checking: Using the Gram correspondence, can you get another algorithm for free?
Transference of Error Bounds
The Gram correspondence also allows us to transfer error bounds between projection approximations and Nyström approximations. Let’s begin with the simplest manifestation of this principle:
Transference of Error Bounds I: Let be a Gram square root of , and let and be the corresponding projection and Nyström approximations generated using the same test matrix . Then
![\[\norm{B - \hat{B}}_{\rm F}^2 = \tr(A - \hat{A}).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1e2e33e007787db8ac81b5521705652c_l3.png "Rendered by QuickLaTeX.com")
This shows that the Frobenius norm error of a projection approximation and the trace error of a Nyström approximation are the same. Using this result, we can immediately transfer error bounds for projection approximations to Nyström approximations and visa versa. For instance, in this previous post, we proved the following error bound for the rank- randomized SVD (with a Gaussian test matrix ):
![\[\expect \norm{B - \hat{B}}_{\rm F}^2 \le \min_{r\le k-2} \left(1 + \frac{r}{k-(r+1)} \right) \norm{B - \lowrank{B}_r}_{\rm F}^2,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1323433654642dc38b1335a77469553e_l3.png "Rendered by QuickLaTeX.com")
Here,

denotes the
best rank-

approximation to
. This result shows that the error of the rank-
randomized SVD is comparable to the best rank-
approximation for any

. Using the transference principle, we immediately obtain a corresponding bound for rank-
Nyström approximation (with a Gaussian test matrix
):
![\[\expect \tr(A - \hat{A}) \le \min_{r\le k-2} \left(1 + \frac{r}{k-(r+1)} \right) \tr(A - \lowrank{A}_r).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5407074bdbcdd0a7918d506ac605b306_l3.png "Rendered by QuickLaTeX.com")
With no additional work, we have converted an error bound for the randomized SVD to an error bound for single-pass randomized Nyström approximation!
For those interested, one can extend the transference of error bounds to general unitarily invariant norms. See the following optional section for details:
Transference of Error Bounds for General Unitarily Invariant Norms
We begin by recalling the notions of (quadratic) unitarily invariant norms; see
this section of a previous post for a longer refresher. A norm

is
unitarily invariant if
![\[\norm{UCV}_{\rm UI} = \norm{C}_{\rm UI}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6911f19fc91e0e3556bdb09361531e6c_l3.png "Rendered by QuickLaTeX.com")
for every matrix

and orthogonal matrices
and

.
Examples of unitarily invariant norms include the nuclear norm

, the Frobenius norm

, and the spectral norm

. (It is not coincidental that all these unitarily invariant norms can be expressed only in terms of the
singular values 
of the matrix
.) Associated to every unitarily invariant norm it its associated
quadratic unitarily invariant norm, defined as
![\[\norm{C}_{\rm Q}^2 = \norm{C^\top C}_{\rm UI} = \norm{CC^\top}_{\rm UI}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f627dafc92609fec4bfd79b7449235f3_l3.png "Rendered by QuickLaTeX.com")
For example,
- The quadratic unitarily invariant norm associated to the nuclear norm
 is the Frobenius norm
is the Frobenius norm  .
.
- The spectral norm is its own associated quadratic unitarily invariant norm
 .
.
This leads us to the more general version of the transference principle:
Transference of Error Bounds II: Let be a Gram square root of , and let and be the corresponding projection and Nyström approximations generated using the same test matrix . Then
![\[\norm{B - \hat{B}}_{\rm Q}^2 = \norm{A - \hat{A}}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2fc3e0434bc399a8e268f4009b4ebe52_l3.png "Rendered by QuickLaTeX.com")
for every unitarily invariant norm and its associated quadratic unitarily invariant norm  .
.
This version of the principle is more general, since the nuclear norm of a psd matrix is the trace, whence
![\[\norm{B - \hat{B}}_{\rm F}^2 = \norm{A - \hat{A}}_* = \tr(A - \hat{A}).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-72b523183dfe27ed53b2d5e9fe5ae56a_l3.png "Rendered by QuickLaTeX.com")
We now also have more examples. For instance, for the spectral norm, we have
![\[\norm{B - \hat{B}}^2 = \norm{A - \hat{A}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8376bea9952904e84ad58071c48665d_l3.png "Rendered by QuickLaTeX.com")
Let us quickly prove the transference of error principle. Write the error  . Thus,
. Thus,
![\[\norm{B - \hat{B}}_{\rm Q}^2 = \norm{(I-\Pi_{B\Omega})B}_{\rm Q}^2 = \norm{B^\top (I-\Pi_{B\Omega})^2B}_{\rm UI}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7a4c680324355da1e53b0d72d315f48c_l3.png "Rendered by QuickLaTeX.com")
The orthogonal projections
and

are equal to their own square, so

Finally, write
and use the Gram correspondence
,
to conclude
![\[\norm{B - \hat{B}}_{\rm Q}^2 = \norm{A - \hat{A}}_{\rm UI},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9e9f3b1f06460a7883813043f8cd7e71_l3.png "Rendered by QuickLaTeX.com")
as desired.
Conclusion
This has been a long post about a simple idea. Many of the low-rank approximation algorithms in the literature output a special type of approximation, either a projection approximation or a Nyström approximation. As this post has shown, these two types of approximation are equivalent, with algorithms and error bounds for one type of approximation transferring immediately to the other format. This equivalence is a powerful tool for the algorithm designer, allowing us to discover new algorithms from old ones, like randomly pivoted Cholesky from randomly pivoted QR.
 of two positive semidefinite matrices is also positive semidefinite. This post will present every proof I know for this theorem, and I intend to edit it to add additional proofs if I learn of them. (Please reach out if you know another!) My goal in this post is to be short and sweet, so I will assume familiarity with many properties for positive semidefinite matrices.
of two positive semidefinite matrices is also positive semidefinite. This post will present every proof I know for this theorem, and I intend to edit it to add additional proofs if I learn of them. (Please reach out if you know another!) My goal in this post is to be short and sweet, so I will assume familiarity with many properties for positive semidefinite matrices. is positive semidefinite (psd, for short) if it is symmetric and satisfies
is positive semidefinite (psd, for short) if it is symmetric and satisfies  for all vectors
for all vectors  . All matrices in this post are real, though the proofs we’ll consider also extend to complex matrices. The entrywise product will be denoted
. All matrices in this post are real, though the proofs we’ll consider also extend to complex matrices. The entrywise product will be denoted  and is defined as
and is defined as  . The entrywise product is also known as the Hadamard product or Schur product.
. The entrywise product is also known as the Hadamard product or Schur product. :
: ![\[x^\top (A\circ M)x = \sum_{i,j=1}^n x_i (A\circ M)_{ij} x_j = \sum_{i,j=1}^n x_i A_{ij} M_{ij} x_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ca498529303f53df914b6597120a87ba_l3.png "Rendered by QuickLaTeX.com")
 , and repackage it as a trace
, and repackage it as a trace ![\[x^\top (A\circ M)x = \sum_{i,j=1}^n x_i A_{ij} x_j M_{ji} = \tr(\operatorname{diag}(x) A \operatorname{diag}(x) M).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a90d263429e7e03a177a6cf648480ca_l3.png "Rendered by QuickLaTeX.com")
 . Substituting these expressions in the trace formula and invoking the cyclic property of the trace, we get
. Substituting these expressions in the trace formula and invoking the cyclic property of the trace, we get ![\[x^\top (A\circ M)x = \tr(\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top C) = \tr(C\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f2629a49866cdb694ccd9c49070a5e6b_l3.png "Rendered by QuickLaTeX.com")
![\[C\operatorname{diag}(x) B^\top B \operatorname{diag}(x) C^\top = G^\top G \quad \text{for } G = B \operatorname{diag}(x) C^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9db0420811bd26d88480f5007736326d_l3.png "Rendered by QuickLaTeX.com")
![\[x^\top (A\circ M)x = \tr(G^\top G) \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a825979a976eeb8f0c30d22ba4b3ef54_l3.png "Rendered by QuickLaTeX.com")
 for every vector
for every vector  , so
, so  and
and  denote the
denote the  th rows of
th rows of ![\[A = \sum_i b_ib_i^\top \quad \text{and} \quad M = \sum_j c_jc_j^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-15aaf1258f117b7a7725a83734ff37fe_l3.png "Rendered by QuickLaTeX.com")
![\[A\circ M = \sum_{i,j} (b_ib_i^\top \circ c_jc_j^\top).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-90054b33d9269313d03d42b53f9ee6fe_l3.png "Rendered by QuickLaTeX.com")
 and
and  is, by direct computation,
is, by direct computation,  . Thus,
. Thus, ![\[A\circ M = \sum_{i,j} (b_i\circ c_j)(b_i\circ c_j)^\top\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bb4a32b8aa99a935e877ef794c60be72_l3.png "Rendered by QuickLaTeX.com")
 be independent random vectors with zero mean and covariance matrices
be independent random vectors with zero mean and covariance matrices  is seen to have zero mean as well. Thus, the
is seen to have zero mean as well. Thus, the  entry of the covariance matrix
entry of the covariance matrix  of
of ![\[\expect[x_iy_ix_jy_j] = \expect[x_ix_j] \expect[y_iy_j] = A_{ij} M_{ij} = (A\circ M)_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-725429d72314bf6d82647383bdfd6035_l3.png "Rendered by QuickLaTeX.com")
 have replaced by expectations
have replaced by expectations ![A = \expect [xx^\top]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ffab58ec6df7d98125e312fa1fc59784_l3.png "Rendered by QuickLaTeX.com") .
.
 of two psd matrices is psd. The entrywise product
of two psd matrices is psd. The entrywise product ![\[A\circ M = ((A\otimes M)_{(i+n(i-1))(i+n(i-1))} : i = 1,\ldots,n).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eeb4d907792549db0d2070858208ab54_l3.png "Rendered by QuickLaTeX.com")
![\[x = \operatorname*{argmin}_{x \in \real^n} \norm{b-Ax}, \quad A \in \real^{m\times n}, \quad b \in \real^m. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5e33407ef09afbe894c4444200ef1529_l3.png "Rendered by QuickLaTeX.com")
 be a sketching matrix for
be a sketching matrix for  of distortion
of distortion  (see these
(see these ![\[(1-\eta)\norm{y} \le \norm{Sy} \le (1+\eta)\norm{y} \quad \text{for every } y \in \operatorname{range}(\onebytwo{A}{b}). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-35e4ad025e13422c8dcab0493f18e7ca_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{x} = \operatorname*{argmin}_{\hat{x}\in\real^n} \norm{Sb-(SA)\hat{x}}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f670046ddc52cc7307dadb6328f2b99c_l3.png "Rendered by QuickLaTeX.com")
 of the sketch-and-solve solution? Here’s a one bound:
of the sketch-and-solve solution? Here’s a one bound: bound). The sketch-and-solve solution (3) satisfies the bound
bound). The sketch-and-solve solution (3) satisfies the bound ![\[\norm{b-A\hat{x}} \le \frac{1+\eta}{1-\eta} \cdot \norm{b-Ax} = (1 + 2\eta + \order(\eta^2))\norm{b-Ax}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d383ca05866d84cdadae5a76138ad6ea_l3.png "Rendered by QuickLaTeX.com")
 is in the range of
is in the range of ![\[(1-\eta) \norm{b-A\hat{x}} \le \norm{S(b-A\hat{x})}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6fcf9e9cb31a6ade0b344486e45944a5_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{b-A\hat{x}} \le \frac{1}{1-\eta}\norm{S(b-A\hat{x})}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cab310db6a63fcdf247bc9d23e5de088_l3.png "Rendered by QuickLaTeX.com")
 is minimized for the value
is minimized for the value  . Thus, its value can only increase by replacing
. Thus, its value can only increase by replacing ![\[\norm{b-A\hat{x}} \le \frac{1}{1-\eta}\norm{S(b-A\hat{x})}\le \frac{1}{1-\eta}\norm{S(b-Ax)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9953de6bf8c5bcfce12c6bdf3c194d0e_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{b-A\hat{x}} \le \frac{1}{1-\eta}\norm{S(b-A\hat{x})}\le \frac{1}{1-\eta}\norm{S(b-Ax)}\le \frac{1+\eta}{1-\eta}\cdot \norm{b-Ax}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-75403516026cfbac74ae561ebf4621b6_l3.png "Rendered by QuickLaTeX.com")
 larger than the minimal least-squares residual
larger than the minimal least-squares residual  . Interestingly, this conclusion is not sharp. In fact, the residual for sketch-and-solve can only be at most
. Interestingly, this conclusion is not sharp. In fact, the residual for sketch-and-solve can only be at most  larger than optimal. This fact has been known at least since
larger than optimal. This fact has been known at least since ![\[A = UC \quad \text{for } U\in\real^{m\times n}, \: B \in \real^{n\times n} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-358dc6a61b3a7b7d9d14b3ac8f72e63e_l3.png "Rendered by QuickLaTeX.com")
![\[r = b-Ax\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3c4e40356c4fb9ea105bf6ac257f8467_l3.png "Rendered by QuickLaTeX.com")
![\[\overline{r} = \frac{r}{\norm{r}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a89ebe07436c947c0a5096796cbd32d4_l3.png "Rendered by QuickLaTeX.com")
 . Consequently,
. Consequently, ![\[\onebytwo{U}{\overline{r}} \quad \text{is an orthonormal basis for } \operatorname{range}(\onebytwo{A}{b}). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3b9498419e5d8ece684c68661d0392be_l3.png "Rendered by QuickLaTeX.com")
 is an orthonormal basis for
is an orthonormal basis for  . To get a sharper analysis of sketch-and-solve, we will need the following result, which shows that
. To get a sharper analysis of sketch-and-solve, we will need the following result, which shows that  is an “almost orthonormal basis”.
is an “almost orthonormal basis”.![\[\sigma_{\rm min}(S\onebytwo{U}{\overline{r}}) \ge 1-\eta, \quad \sigma_{\rm max}(S\onebytwo{U}{\overline{r}}) \le 1+\eta. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d7368f84e4ca7f8cb2432c52ef790c9a_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\onebytwo{U}{\overline{r}}^\top S^\top S\onebytwo{U}{\overline{r}} - I} \le 2\eta+\eta^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5b215924bb1be4cf7ca3411f876fceb5_l3.png "Rendered by QuickLaTeX.com")
![\[\sigma_{\rm min}(S\onebytwo{U}{\overline{r}}) = \min_{\norm{z}=1} \norm{S\onebytwo{U}{\overline{r}}z}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6008f71b85c63a98925efa07829b3bd6_l3.png "Rendered by QuickLaTeX.com")
 is in the range of
is in the range of ![\[\sigma_{\rm min}(S\onebytwo{U}{\overline{r}}) = \min_{\norm{z}=1} \norm{S\onebytwo{U}{\overline{r}}z} \ge (1-\eta) \min_{\norm{z}=1} \norm{\onebytwo{U}{\overline{r}}z} = 1-\eta.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ea84e76120e01c72b6c9e437203f8a6e_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\onebytwo{U}{\overline{r}}z} = \norm{z} \quad \text{for every } z \in \real^{n+1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8571bbac885d1b2325af503df4fe077c_l3.png "Rendered by QuickLaTeX.com")
 are equal to the squared singular values of
are equal to the squared singular values of  gives
gives ![\[(1-\eta)^2 \le \lambda_{\rm min}(\onebytwo{U}{\overline{r}}^\top S^\top S\onebytwo{U}{\overline{r}}) \le \lambda_{\rm max}(\onebytwo{U}{\overline{r}}^\top S^\top S\onebytwo{U}{\overline{r}}) \le (1+\eta)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4401a9cb714816c7b57988754c1b9ba4_l3.png "Rendered by QuickLaTeX.com")
 :
:![\[-2\eta+\eta^2 \le \lambda_{\rm min}(\onebytwo{U}{\overline{r}}^\top S^\top S\onebytwo{U}{\overline{r}}-I) \le \lambda_{\rm max}(\onebytwo{U}{\overline{r}}^\top S^\top S\onebytwo{U}{\overline{r}}-I) \le 2\eta+\eta^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f4952e12d4ba5d4bdae987806486e89d_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\onebytwo{U}{\overline{r}}^\top S^\top S\onebytwo{U}{\overline{r}}-I} \le \max(2\eta+\eta^2,-(-2\eta+\eta^2)) = 2\eta+\eta^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c07ba0eedf5c8149783a561cc9817845_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{b-A\hat{x}}^2 \le \left(1 + \frac{(2\eta+\eta^2)^2}{(1-\eta)^4}\right) \norm{b-Ax}^2 \le \left(1 + \frac{9\eta^2}{(1-\eta)^4}\right)\norm{b-Ax}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e8f5d6e0c0bb7ce2f60a21b7ee654ab8_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{b-A\hat{x}}^2 \le (1+4\eta^2+\order(\eta^3)) \norm{b-Ax}^2 \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-65d5e45cb5ec3958371d247a488c150f_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{b-A\hat{x}} \le (1+2\eta^2+\order(\eta^3)) \norm{b-Ax}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3772f3871e0fcf2d67f39b1931487e3f_l3.png "Rendered by QuickLaTeX.com")
 :
: ![\[b - A\hat{x} = b - Ax + Ax - A\hat{x} = r + A(\hat{x}-x).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e233c900f3f02cbc4b418b0e5c4aebd9_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{b-A\hat{x}}^2 = \norm{r}^2 + \norm{A(\hat{x}-x)}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4b9f6e908d43660adf87d2681ce79b6c_l3.png "Rendered by QuickLaTeX.com")
 , it will help us to have a more convenient formula for
, it will help us to have a more convenient formula for  . To this end, reparametrize the sketch-and-solve least-squares problem as an optimization problem over the error
. To this end, reparametrize the sketch-and-solve least-squares problem as an optimization problem over the error  :
:![\[\hat{x} = \operatorname*{argmin}_{\hat{x}\in\real^n} \norm{S(b - Ax)-SA(\hat{x}-x)} \implies \hat{x} - x = \operatorname*{argmin}_{e\in\real^n} \norm{Sr - SAe}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a29317b3e6eaf6fab249be69d62032ee_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{x} - x = (A^\top S^\top SA)^{-1} A^\top S^\top Sr.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-33e6648bec41d7a892221020eac0fd31_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{x} - x = C^{-1} (U^\top S^\top SU)^{-1} U^\top S^\top Sr.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-29d0f4c0bb950c85d705aae8d6222a3c_l3.png "Rendered by QuickLaTeX.com")
![\[A(\hat{x} - x) = U(U^\top S^\top SU)^{-1} U^\top S^\top Sr.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-50557b5767040195d4368556be2cc235_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain ![\[\norm{A(\hat{x} - x)} \le \norm{(U^\top S^\top S U)^{-1}} \cdot \norm{U^\top S^\top S \overline{r}} \cdot \norm{r}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c1dea39a12d631bbf5066c326b2abd12_l3.png "Rendered by QuickLaTeX.com")
 is a submatrix of
is a submatrix of ![\[\sigma_{\rm min}(SU) \ge \sigma_{\rm min}(S\onebytwo{U}{\overline{r}}) \ge 1-\eta.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5496deb862aff64991febf9b5e209a13_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{(U^\top S^\top S U)^{-1}} = \sigma_{\rm min}^2(SU) \le \frac{1}{(1-\eta)^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b57471b3a2b8e33eb581bc2108b4a58f_l3.png "Rendered by QuickLaTeX.com")
 is a submatrix of
is a submatrix of  . Thus, by (8),
. Thus, by (8), ![\[\norm{U^\top S^\top S \overline{r}} \le \norm{\onebytwo{U}{\overline{r}}S^\top S\onebytwo{U}{\overline{r}} - I}\le 2\eta + \eta^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-147f6cc80507de0d27d6df847ed86a7e_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{A(\hat{x} - x)} \le \frac{2\eta+\eta^2}{(1-\eta)^2} \cdot \norm{r}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-430bcb217499f227f5cec8ca1d3afd5a_l3.png "Rendered by QuickLaTeX.com")
 , Theorem 3 identifies the correct
, Theorem 3 identifies the correct  Gaussian matrix is an
Gaussian matrix is an  . The expected least-squares residual is
. The expected least-squares residual is ![\[\expect \norm{b - A\hat{x}}^2 \approx \left(1 + \frac{\eta^2}{(1+\eta)(1-\eta)}\right) \norm{b - Ax}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c095e0b726aeb7fadf644a4a9efdda18_l3.png "Rendered by QuickLaTeX.com")
 in the limit
in the limit  , whereas the bound (14) for Gaussian embeddings scales like
, whereas the bound (14) for Gaussian embeddings scales like  . We leave it as a conjecture/open problem whether there is an improved argument for general subspace embeddings:
. We leave it as a conjecture/open problem whether there is an improved argument for general subspace embeddings:![\[\norm{b-A\hat{x}}^2 \le \left(1 + \frac{C\eta^2}{1-\eta}\right)\norm{b-Ax}^2\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6d46a32b59fd74bc2000b5df173175e1_l3.png "Rendered by QuickLaTeX.com")
 .
.
 scaling for a different type of dimensionality reduction (leverage score sampling), check out
scaling for a different type of dimensionality reduction (leverage score sampling), check out  be vectors, assemble the matrix
be vectors, assemble the matrix  , and form the
, and form the ![\[A = B^\top B = \onebytwo{x}{y}^\top \onebytwo{x}{y} = \twobytwo{\norm{x}^2}{y^\top x}{x^\top y}{\norm{y}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3da148e3bfb2685780c2ac01a05e4c2a_l3.png "Rendered by QuickLaTeX.com")
![\[\det(A) = \norm{x}^2\norm{y}^2 - |x^\top y|^2 \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-93675ca9816700c7f444d9c54aef2b09_l3.png "Rendered by QuickLaTeX.com")
![\[|x^\top y| \le \norm{x}\norm{y}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bf031db9e34714a3b017164922c25253_l3.png "Rendered by QuickLaTeX.com")
 be strictly positive numbers. Then the inverse of their average is no bigger than the average of their inverses:
be strictly positive numbers. Then the inverse of their average is no bigger than the average of their inverses: ![\[\left( \frac{1}{n} \sum_{i=1}^n x_i \right)^{-1} \le \frac{1}{n} \sum_{i=1}^n \frac{1}{x_i}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff75b76509202932a39a33e6748ba18c_l3.png "Rendered by QuickLaTeX.com")
 into a
into a  positive semidefinite matrix
positive semidefinite matrix  . Taking the average of all such matrices, we observe that
. Taking the average of all such matrices, we observe that ![\[A = \twobytwo{\frac{1}{n} \sum_{i=1}^n x_i}{1}{1}{\frac{1}{n} \sum_{i=1}^n \frac{1}{x_i}}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f5bbf22f522171bdf7e178ce8030634a_l3.png "Rendered by QuickLaTeX.com")
![\[\det(A) = \left(\frac{1}{n} \sum_{i=1}^n x_i\right) \left(\frac{1}{n} \sum_{i=1}^n \frac{1}{x_i}\right) - 1 \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8c87d66274b7a87984791af241e45f9e_l3.png "Rendered by QuickLaTeX.com")
 is interpreted as the
is interpreted as the  . The matrix square root
. The matrix square root  . Moreover, it is the unique matrix with these properties.
. Moreover, it is the unique matrix with these properties. , where
, where  is a matrix with orthonormal columns and
is a matrix with orthonormal columns and  is an upper trapezoidal matrix, up to a permutation of the rows. This factorized form is easy to compute roughly following steps 1–3 above, which explains why we call that procedure a “
is an upper trapezoidal matrix, up to a permutation of the rows. This factorized form is easy to compute roughly following steps 1–3 above, which explains why we call that procedure a “ for a weight matrix
for a weight matrix  . This type of factorization is known as an
. This type of factorization is known as an  , this simplifies to
, this simplifies to  The matrix
The matrix  , leading the second and third term to vanish. Finally, using the relation
, leading the second and third term to vanish. Finally, using the relation 
 as output.
as output.
 is nonnegative and satisfies
is nonnegative and satisfies  if and only if
if and only if  ; these statements justify why
; these statements justify why  is an appropriate expression for measuring the error of the approximation
is an appropriate expression for measuring the error of the approximation  .
. . This identity, too, follows by the transference principle, since
. This identity, too, follows by the transference principle, since  are, themselves, projection approximations and Nyström approximations corresponding to choosing
are, themselves, projection approximations and Nyström approximations corresponding to choosing  , whose dimensions will be
, whose dimensions will be  throughout this post. Beginning from a trivial initial solution
throughout this post. Beginning from a trivial initial solution  , the method works by repeating the following two steps for
, the method works by repeating the following two steps for  :
: of
of ![\[\prob\{ i_t = j\} = \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-10df9446203ebd72722321adfe647007_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  th row of
th row of  onto the solution space of the equation
onto the solution space of the equation  , obtaining
, obtaining  .
. satisfying
satisfying  ), RK is
), RK is ![\[\expect\left[ \norm{x_t - x_\star}^2 \right] \le (1 - \kappa_{\rm dem}^{-2})^t \norm{x_\star}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eee339c47be34bc0343976d0b6383dd5_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem} = \frac{\norm{A}_{\rm F}}{\sigma_{\rm min}(A)} = \sqrt{\sum_i \left(\frac{\sigma_i(A)}{\sigma_{\rm min}(A)}\right)^2} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6fbef84dde9dc3a326a898ad4e6cc0a2_l3.png "Rendered by QuickLaTeX.com")
 are the
are the  , where
, where  is the
is the  is the
is the  , so it takes roughly
, so it takes roughly  row accesses to reduce the error by a constant factor. Compare this to gradient descent, which requires
row accesses to reduce the error by a constant factor. Compare this to gradient descent, which requires  row accesses.
row accesses. , which can be expressed using the
, which can be expressed using the  .
.![\[x_\star = \operatorname{argmin}_{x \in \real^d} \norm{b - Ax}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c13b23b7fa8f955b4410fc6c09e27f73_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\mathbb{E}[x_t] - x_\star}^2 \le (1 - \kappa_{\rm dem}^{-2})^{2t} \norm{x_\star}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ab1fa6069874cae173b07c6dd911e972_l3.png "Rendered by QuickLaTeX.com")
 .
. which could then be averaged together. This approach is inefficient as each solution
which could then be averaged together. This approach is inefficient as each solution  is computed separately.
is computed separately. , chosen so that the bias
, chosen so that the bias ![\norm{\expect[x_{t_{\rm b}}] - x_\star}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f4d9818474fef0214cae964e961ff41d_l3.png "Rendered by QuickLaTeX.com") is small. For each
is small. For each  ,
,  is a nearly unbiased approximation to the least-squares solution
is a nearly unbiased approximation to the least-squares solution ![\[\overline{x}_t = \frac{x_{t_{\rm b} +1} + x_{t_{\rm b}+2} + \cdots + x_t}{t-t_{\rm b}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-704f302ecc7ffe66be1c58b43aff4f13_l3.png "Rendered by QuickLaTeX.com")
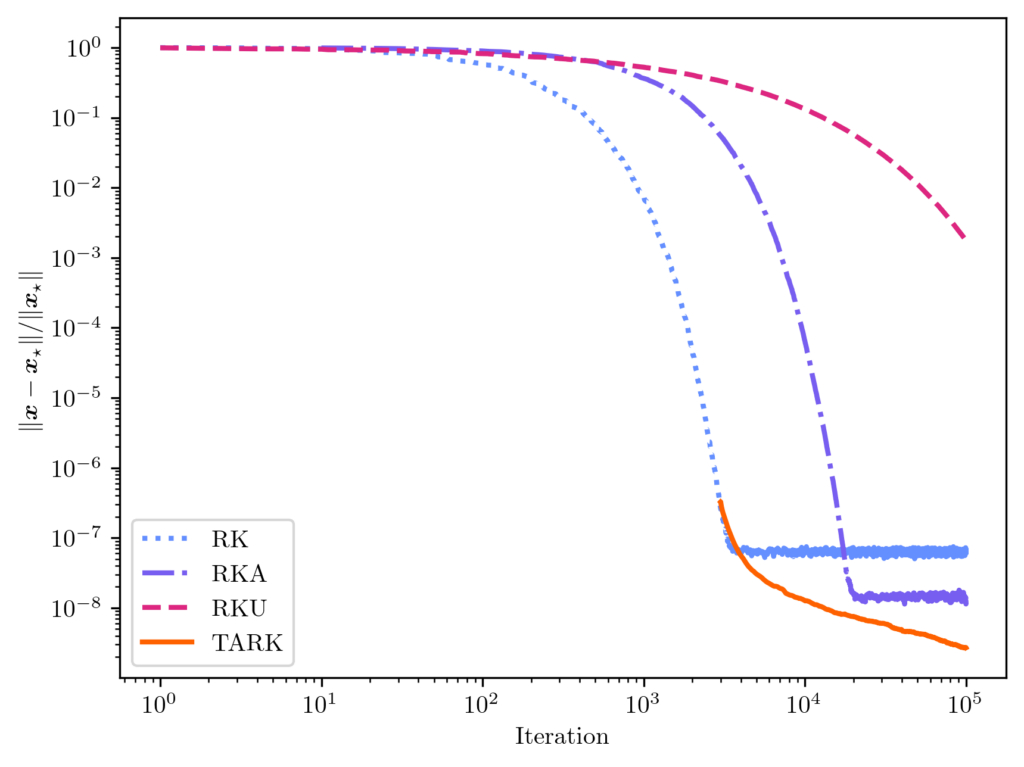
 is the tail-averaged randomized Kaczmarz (TARK) estimator. By Theorem 1, we know the TARK estimator has an exponentially small bias:
is the tail-averaged randomized Kaczmarz (TARK) estimator. By Theorem 1, we know the TARK estimator has an exponentially small bias: ![\[\norm{\expect[\overline{x}_t] - x_\star} \le (1 - \kappa_{\rm dem}^{-2})^{2(t_{\rm b}+1)} \norm{x_\star}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8462c91ce74898bbdf8f65e477ff3f7c_l3.png "Rendered by QuickLaTeX.com")
![\[\expect [\norm{\overline{x}_t - x_\star}^2] \le (1-\kappa_{\rm dem}^{-2})^{t_{\rm b}+1} \norm{x_\star}^2 + \frac{2\kappa_{\rm dem}^4}{t-t_{\rm b}} \frac{\norm{b-Ax_\star}^2}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2a8b02cbb1aaca3e89c4671b2fda1c37_l3.png "Rendered by QuickLaTeX.com")
 . While the Monte Carlo rate of convergence may be unappealing,
. While the Monte Carlo rate of convergence may be unappealing,  ;
;  . We found this underrelaxation parameter to lead to a smaller error than the other popular underrelaxation parameter schedule
. We found this underrelaxation parameter to lead to a smaller error than the other popular underrelaxation parameter schedule  .
.
![\[\prob \{ i_t = j \} = \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2} \eqqcolon p^{\rm RK}_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1d6a18d4d7f13a01b6598d317ed4d10e_l3.png "Rendered by QuickLaTeX.com")
 .
.
 .
. :
: ![\[\prob \{i_t = j\} = p_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c65a3dab2b76666bb80f4b9ba9eba61c_l3.png "Rendered by QuickLaTeX.com")
![\[D \coloneqq \diag\left( \sqrt{\frac{p_j}{p_j^{\rm RK}}} : j =1,\ldots,n\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c4a48ae205003d974f3d9c22f05b4971_l3.png "Rendered by QuickLaTeX.com")
 is equivalent to the standard RK algorithm run on the diagonally reweighted least-squares problem
is equivalent to the standard RK algorithm run on the diagonally reweighted least-squares problem ![\[x_{\rm weighted} = \argmin_{x\in\real^d} \norm{Db-(DA)x}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c8f0cfed11c57aecc99422efa2e26145_l3.png "Rendered by QuickLaTeX.com")
 rather than the original least-squares solution
rather than the original least-squares solution  [mfn}Note that, for this experiment we represent the polynomial
[mfn}Note that, for this experiment we represent the polynomial  using its monomial coefficients
using its monomial coefficients  , which has
, which has  equispaced points. We compare the unweighted least-squares solution (orange solid curve) to the weighted least-squares solution using uniform RK weights
equispaced points. We compare the unweighted least-squares solution (orange solid curve) to the weighted least-squares solution using uniform RK weights  (blue dash-dotted curve). These two curves differ meaningfully, with the weighted least-squares solution having higher error at the ends of the interval but more accuracy in the middle. These differences can be explained looking at the weights (diagonal entries of
(blue dash-dotted curve). These two curves differ meaningfully, with the weighted least-squares solution having higher error at the ends of the interval but more accuracy in the middle. These differences can be explained looking at the weights (diagonal entries of  , grey dotted curve), which are lower at the ends of the interval than in the center.
, grey dotted curve), which are lower at the ends of the interval than in the center.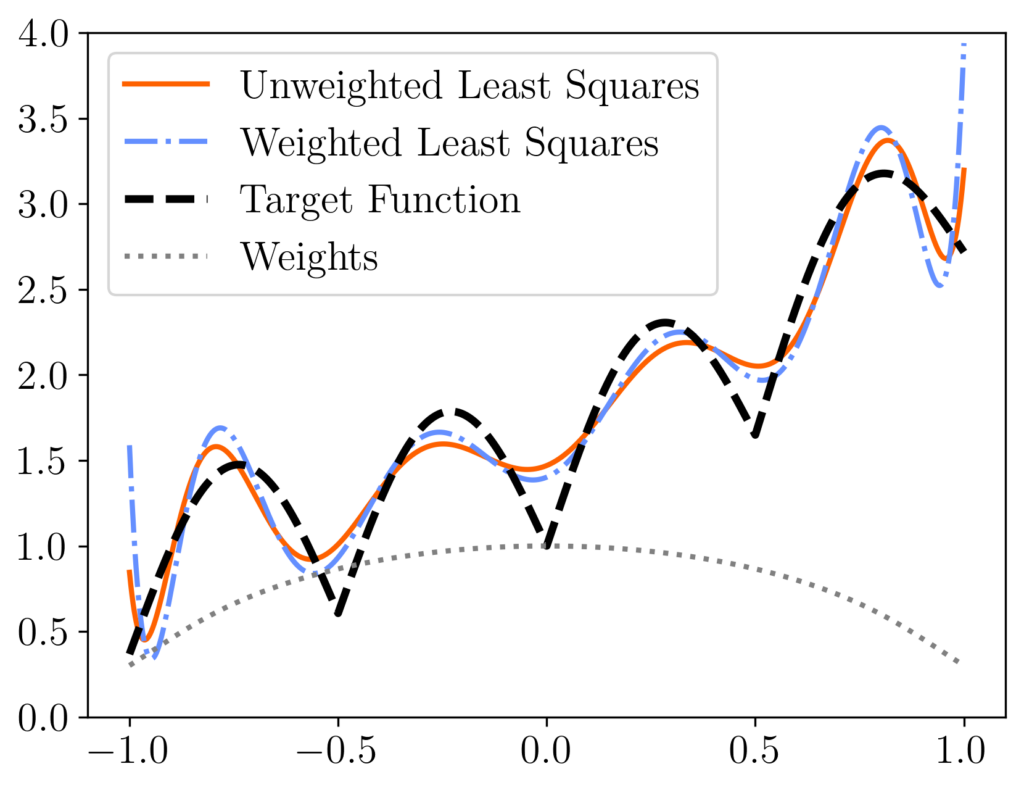
![\[x_{t+1} = \left( I - \frac{a_{i_t}^{\vphantom{\top}}a_{i_t}^\top}{\norm{a_{i_t}}^2} \right)x_t + \frac{b_{i_t}a_{i_t}}{\norm{a_{i_t}}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ebdcce812d0667315ce96a9a10076256_l3.png "Rendered by QuickLaTeX.com")
 denote the
denote the ![\begin{align*}\expect_{i_t}[x_{t+1}] &= \sum_{j=1}^n \left[\left( I - \frac{a_j^{\vphantom{\top}} a_j^\top}{\norm{a_j}^2} \right)x_t + \frac{b_ja_j}{\norm{a_j}^2}\right] \prob\{i_t=j\}\\ &=\sum_{j=1}^n \left[\left( \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2}I - \frac{a_j^{\vphantom{\top}} a_j^\top}{\norm{A}_{\rm F}^2} \right)x_t + \frac{b_ja_j}{\norm{A}_{\rm F}^2}\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6313b1e410f036a4151bb5c5f6cb89c8_l3.png "Rendered by QuickLaTeX.com")
 and
and  directly. Therefore, we obtain
directly. Therefore, we obtain ![\[\expect_{i_t}[x_{t+1}] = \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right) x_t + \frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5d568d3ca43156f34ddcb4556ac18c68_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[x_{t+1}] = \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right) \expect[x_t] + \frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0ac2ab733dcde2fc5c33d3d1b668523c_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain ![\[\expect[x_t] = \left[\sum_{i=0}^{t-1} \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i \right]\cdot\frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-320f299e0726caf09895b9ce10722c65_l3.png "Rendered by QuickLaTeX.com")
![\expect[x_t]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-41d92b12c75d466d126bae8b5af9c2a5_l3.png "Rendered by QuickLaTeX.com") using a matrix
using a matrix  satisfies the formula
satisfies the formula ![\[\sum_{i=0}^\infty y^i = (1-y)^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bb3e56cc5e0bc7e9f8e5eedf9f14cf68_l3.png "Rendered by QuickLaTeX.com")
 , we get
, we get ![\[\sum_{i=0}^\infty (1-x)^i = x^{-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a7051c9c16502097cf3a3bab37487727_l3.png "Rendered by QuickLaTeX.com")
 . With a little effort, one can check that the same formula
. With a little effort, one can check that the same formula![\[\sum_{i=0}^\infty (I-X)^i = X^{-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b0bc6ad83e00c955148a0ecb1bba1de5_l3.png "Rendered by QuickLaTeX.com")
 satisfying
satisfying  . These conditions hold for the matrix
. These conditions hold for the matrix  since
since ![\[\left(\frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^{-1} = \sum_{i=0}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6b584a1bef1b7a5776457988bb29d3b7_l3.png "Rendered by QuickLaTeX.com")
![\[\left(\frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^{-1} = \sum_{i=0}^{t-1} \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i + \sum_{i=t}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-000ddcb9b9d9b6ee0fc32c784f7348c7_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain ![\[\expect[x_t] = x_\star - \left[\sum_{i=t}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i \right]\cdot\frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6400bfb8abb56d715838ee2386b5ebd2_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[x_t] - x_\star = - \left(I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^t \left[\sum_{i=0}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i \right]\cdot\frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8d61b65cfb176614048ae1b5976d47bf_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[x_t] - x_\star = - \left(I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^t x_\star.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4deff646d7b889bd0c37907ea6a64dbb_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\expect[x_t] - x_\star}^2 \le \norm{I - \frac{A^\top A}{\norm{A}_{\rm F}^2}}^{2t} \norm{x}_\star^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cd024bd8769d75cbb989c08c3d4834da_l3.png "Rendered by QuickLaTeX.com")
 . Let
. Let  be a (
be a ( . Then
. Then ![\[I - \frac{A^\top A}{\norm{A}_{\rm F}^2} = I - V \cdot\frac{\Sigma^2}{\norm{A}_{\rm F}^2} \cdot V^\top = V \left( I - \frac{\Sigma^2}{\norm{A}_{\rm F}^2}\right)V^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b9d13c9f13a4e81778d6d48f25d891e5_l3.png "Rendered by QuickLaTeX.com")
![\[I - \Sigma^2/\norm{A}_{\rm F}^2 = \diag(1 - \sigma^2_{\rm max}(A)/\norm{A}_{\rm F}^2,\ldots,1-\sigma_{\rm min}^2(A)/\norm{A}_{\rm F}^2).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8e2959ad366626763fb9ead487096980_l3.png "Rendered by QuickLaTeX.com")
 . We have invoked the definition of the Demmel condition number (2). Therefore, plugging into (7), we obtain
. We have invoked the definition of the Demmel condition number (2). Therefore, plugging into (7), we obtain ![\[\norm{\expect[x_t] - x_\star}^2 \le \left(1-\kappa_{\rm dem}^{-2}\right)^{2t} \norm{x}_\star^2,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-87d41d4e2aee6259779d71c186f9fc9a_l3.png "Rendered by QuickLaTeX.com")
 an
an  ?
? . Throughout this post, I will assume knowledge of sketching; see my previous
. Throughout this post, I will assume knowledge of sketching; see my previous  , and these parameters are related
, and these parameters are related  .
.![\[\expect\Big[\big((SA)^\top (SA)\big)^{-1}\Big] \ne \big(A^\top A\big)^{-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-16fac3fccb2c5056d7f3ab930c64e5bc_l3.png "Rendered by QuickLaTeX.com")
 . The sketch-and-solve solution
. The sketch-and-solve solution  does not change under a scaling of the matrix
does not change under a scaling of the matrix ![\[S \in \real^{\ell\times n} \quad \text{with } S_{ij} \sim \text{Normal}(0,1) \text{ iid},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3776da0da83625d5c9a60ed2d1a89d65_l3.png "Rendered by QuickLaTeX.com")
 be a full column-rank matrix. Then
be a full column-rank matrix. Then ![\[\expect[(SA)^\dagger(Sb)] = A^\dagger b. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d55b038f9d67957e7faedf7368fde72_l3.png "Rendered by QuickLaTeX.com")
 is an unbiased estimate for the least-squares solution
is an unbiased estimate for the least-squares solution  .
.
![\[A^\dagger b = \operatorname*{argmin}_{x\in\real^d} \norm{Ax - b}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d4040378ee11d2e536aecc07ead56b7f_l3.png "Rendered by QuickLaTeX.com")
![\[A^\dagger = (A^\top A)^{-1} A^\top,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e59713cce775d9405d66c90623c5f8ae_l3.png "Rendered by QuickLaTeX.com")
 has the same distribution as
has the same distribution as ![\[A = U\twobyone{\Sigma}{0}V^\top\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6bdf747d3ed1ce328426522c9fe86cfa_l3.png "Rendered by QuickLaTeX.com")
![\[A \mapsto U^\top A V, \quad S \mapsto V^\top SU, \quad b \mapsto U^\top b.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-43261dc6b3a96da26ace4d4b84fa9a44_l3.png "Rendered by QuickLaTeX.com")
![\[A = \twobyone{\Sigma}{0}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4fd9ee2c473a0d1ab80b3bc37e8554bf_l3.png "Rendered by QuickLaTeX.com")
 and
and ![\[b = \twobyone{b_1}{b_2}, \quad S = \onebytwo{S_1}{S_2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-041e66e0d00f617f40c3465a2d47d659_l3.png "Rendered by QuickLaTeX.com")
![\[A^\dagger b = \Sigma^{-1}b_1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3ef2790282b895b475158cd44e0ad67e_l3.png "Rendered by QuickLaTeX.com")
 . Begin by using the normal equations (4) to write out the sketch-and-solve solution
. Begin by using the normal equations (4) to write out the sketch-and-solve solution ![\[(SA)^\dagger (Sb) = [(SA)^\top (SA)]^{-1}(SA)^\top (Sb).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-baee67fa625fe2792e3c468af78b0ec0_l3.png "Rendered by QuickLaTeX.com")
![\[(SA)^\dagger (Sb) = [\Sigma S_1^\top S_1\Sigma]^{-1}\Sigma S_1^\top \onebytwo{S_1}{S_2}\twobyone{b_1}{b_2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1aefd4e03296f2197f1ea8a5d6e585b1_l3.png "Rendered by QuickLaTeX.com")
![\[(SA)^\dagger (Sb) = \Sigma^{-1}(S_1^\top S_1)^{-1} S_1^\top (S_1b_1 + S_2 b_2).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-40cec8d2d08041c37fe07e07014f527d_l3.png "Rendered by QuickLaTeX.com")
![\[(SA)^\dagger (Sb) = \Sigma^{-1}b_1 + \Sigma^{-1} S_1^\dagger S_2b_2 = A^\dagger b + \Sigma^{-1} S_1^\dagger S_2b_2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-24b04e4266015e389aa6015e7bd466e7_l3.png "Rendered by QuickLaTeX.com")
 and
and  are independent and
are independent and ![\expect[S_2] = 0](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-baf5617b019410573ecb50d61a05cbcf_l3.png "Rendered by QuickLaTeX.com") . Thus, taking expectations, we obtain
. Thus, taking expectations, we obtain![\[\expect[(SA)^\dagger (Sb)] = A^\dagger b + \Sigma^{-1} \expect[S_1^\dagger] \expect[S_2]b_2 = A^\dagger b.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5c5a811ba6efc9b4d2a174cd9612e48d_l3.png "Rendered by QuickLaTeX.com")
 columns of
columns of  columns of
columns of ![\[\expect \norm{A\hat{x} - b}^2,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-081e7fd65b359239c4efb6153375a7c4_l3.png "Rendered by QuickLaTeX.com")
 is the sketch-and-solve solution. We write the expected residual norm as
is the sketch-and-solve solution. We write the expected residual norm as ![\[\expect\norm{A\hat{x} - b}^2 = \expect\norm{\twobyone{b_1 + S_1^\dagger S_2b_2}{0} - \twobyone{b_1}{b_2}}^2 = \norm{b_2}^2 + \expect\norm{S_1^\dagger S_2b_2}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6687022eb06ca4c6e3a437f7c61e2155_l3.png "Rendered by QuickLaTeX.com")
![\[\expect\norm{A\hat{x} - b}^2 = \left(1+\frac{d}{\ell-d-1}\right)\norm{b_2}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1a65c0e13fb70224700e2d89b338b550_l3.png "Rendered by QuickLaTeX.com")
 is the optimal least-squares residual
is the optimal least-squares residual  . Thus, we have shown
. Thus, we have shown ![\[\expect\norm{A\hat{x} - b}^2 = \left(1+\frac{d}{\ell-d-1}\right)\min_{x\in\real^d} \norm{Ax-b}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b602470a395855b60b5cd5ee50cddc58_l3.png "Rendered by QuickLaTeX.com")
 larger than the optimal value. In particular,
larger than the optimal value. In particular, ![\[\expect\norm{A\hat{x} - b}^2 = \left(1+\varepsilon\right)\min_{x\in\real^d} \norm{Ax-b}^2 \quad \text{when } \ell = \frac{d}{\varepsilon} + d + 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a708e1047cc1c00320bf8492479bd52_l3.png "Rendered by QuickLaTeX.com")
 be random vectors in
be random vectors in  drawn uniformly at random from the sphere of radius
drawn uniformly at random from the sphere of radius  ,
, -dimensional
-dimensional ![\[q \coloneqq x^\top Ax\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-45122559a9bc50978d4374ac1165c654_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{\tr}\coloneqq \frac{1}{m}\sum_{i=1}^m x_i^\top Ax_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a839d52ecc78895dd6d9dbc78dd5b62_l3.png "Rendered by QuickLaTeX.com")
 and
and  are unbiased estimates for the trace of
are unbiased estimates for the trace of ![\expect[q] = \expect[\hat{\tr}] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1fca19e415f63f68b7b6ecf288acbd80_l3.png "Rendered by QuickLaTeX.com") . The goal of this post will be to bound the probability of these quantities being much smaller or larger than the trace of
. The goal of this post will be to bound the probability of these quantities being much smaller or larger than the trace of  independent copies of the random variable
independent copies of the random variable ![\[\overline{A} = A - \frac{\tr A}{n}I.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b89d7b20195cae2dbb4b24e2f9220fd0_l3.png "Rendered by QuickLaTeX.com")
 has the effect of shifting
has the effect of shifting  has trace zero.
has trace zero.
![\[q = x^\top A x = x^\top \overline{A} x + \frac{\tr A}{n} \cdot x^\top I x = x^\top \overline{A} x + \tr A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cb8b5348af1d8c3c048afba553e988e0_l3.png "Rendered by QuickLaTeX.com")
 . Rearranging, we see that the error
. Rearranging, we see that the error  satisfies
satisfies![\[\overline{q} = q-\tr A = x^\top \overline{A}x.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-59729a383a00f26c1d1986a133833f6c_l3.png "Rendered by QuickLaTeX.com")
 depends only on the centered matrix
depends only on the centered matrix  , and the
, and the  . These observations will be important later.
. These observations will be important later. , define the cumulant generating function (cgf)
, define the cumulant generating function (cgf) ![\[\xi_z(\theta) = \log (\expect [\exp(\theta z)]).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9eb081bc7a576cbb8155e2258db0767c_l3.png "Rendered by QuickLaTeX.com")
 is a vector with
is a vector with  , where
, where  is a scaling factor.
is a scaling factor. is a
is a 
 degrees of freedom.
degrees of freedom.![\expect[a^2] = 1](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3adf3f2ea06eccac656d267691ed9870_l3.png "Rendered by QuickLaTeX.com") .
.![\expect[\norm{g}^2] = \sum_{i=1}^n \expect[g_i^2] = n](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-87fc6bdfd4ff8ef60a06a7d1dfc1a15e_l3.png "Rendered by QuickLaTeX.com") . But also,
. But also, ![\expect[\norm{g}^2] = \expect[a^2] \cdot \expect[\norm{x}^2] = \expect[a^2] \cdot n](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b90c580d42ea4adbaf31a6421a60d6f1_l3.png "Rendered by QuickLaTeX.com") . Therefore,
. Therefore, ![\expect[a^2]=1](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4b3ece1b58b970d5d730faba98e9fd7d_l3.png "Rendered by QuickLaTeX.com") .
.![\[g^\top \overline{A} g = a^2 \cdot x^\top \overline{A} x = a^2 \cdot \overline{q}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-674d36b27b7161eb9ed7ae34d5e142a1_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{\overline{q}}(\theta) \le \xi_{g^\top A g}(\theta) \quad \text{for all } \theta \in \real.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2df1fbd13a1bec6dca240b4ca2767db6_l3.png "Rendered by QuickLaTeX.com")
![\[\xi_{z}(\theta) \le \xi_{bz}(\theta) \quad \text{for all } \theta \in \real.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-837f893d19f64a6e9371cc8dd55350b4_l3.png "Rendered by QuickLaTeX.com")
 and
and  denote expectations take over the randomness in
denote expectations take over the randomness in ![\expect[\cdot] = \expect_z[\expect_b[\cdot ]]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c374e285ed91d07eb4d3be8b7146dee1_l3.png "Rendered by QuickLaTeX.com") . Begin with the right-hand side and invoke
. Begin with the right-hand side and invoke ![\[\xi_{bz}(\theta) = \xi_z(\theta) = \log (\expect_z [\expect_b [\exp(\theta bz)]]) \ge \log (\expect_z [\exp(\theta \expect_b[b]z)]) = \xi_z(\theta).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eee4090aaf6cc65d0c78010a5aadcc78_l3.png "Rendered by QuickLaTeX.com")
![\expect[b] = 1](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d309968b9a6a91bbf504b3813908e619_l3.png "Rendered by QuickLaTeX.com") and the definition of the cgf.
and the definition of the cgf.
 for all
for all  .
. for a trace-zero matrix
for a trace-zero matrix ![\[\xi_{\overline{q}}(\theta) \le \xi_{g^\top \overline{A} g}(\theta) \le \frac{\theta^2 \norm{\overline{A}}_{\rm F}^2}{1 - 2\theta \norm{\overline{A}}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bf41f69447c1db0c3efc7f21316ec906_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{ x^\top A x - \tr(A) \ge t \} \le \exp \left( - \frac{t^2/2}{2 \norm{\overline{A}}_{\rm F}^2 + 2 \overline{\norm{\overline{A}}t}} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-88a3d363a047368765bff1beb2bfca17_l3.png "Rendered by QuickLaTeX.com")
 by instantiating this result with
by instantiating this result with  .
. ![\[\prob \{ |x^\top A x - \tr(A)| \ge t \} \le 2\exp \left( - \frac{t^2/2}{2 \norm{\overline{A}}_{\rm F}^2 + 2 \overline{\norm{\overline{A}}t}} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bb2c0859ac9f678cba1125b7737f7f21_l3.png "Rendered by QuickLaTeX.com")
 or
or  are known as
are known as  for small
for small  and
and  for large
for large  . This pattern of results “subgaussian scaling for small
. This pattern of results “subgaussian scaling for small ![\[\prob \{ g^\top A g - \tr A \ge t \} \le \exp \left( -\frac{t^2/2}{C\norm{A}_{\rm F}^2 + c t \norm{A}}\right)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e705aa2729030afd7f85f60e8e6f55bb_l3.png "Rendered by QuickLaTeX.com")
 . For vectors on the sphere,
. For vectors on the sphere,  and
and  have been replaced by
have been replaced by  and
and  which are always smaller (and sometimes much smaller). The smaller tail probabilities for
which are always smaller (and sometimes much smaller). The smaller tail probabilities for  has tail probabilities roughly of size
has tail probabilities roughly of size  . For small
. For small  . The true variance of
. The true variance of  is
is ![\[\Var(q) = \frac{n}{n+2} \cdot 2\norm{\overline{A}}_{\rm F}^2,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-64f86ad74130c669f06a98a0efa02835_l3.png "Rendered by QuickLaTeX.com")
 easily follow. Indeed, the cgf is additive for independent random variables and satisfies the identity
easily follow. Indeed, the cgf is additive for independent random variables and satisfies the identity  for constant $c, so
for constant $c, so ![\[\xi_{\overline{\tr}}(\theta) = \sum_{i=1}^m \xi_{x_i^\top Ax_i}(\theta/m) \le m \cdot \frac{(\theta/m)^2 \norm{\overline{A}}_{\rm F}^2}{1 - 2(\theta/m)\norm{\overline{A}}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-82d5af32e470db6ff89fd02927bbe65b_l3.png "Rendered by QuickLaTeX.com")

 is a vector. For simplicity, we assume that this system is
is a vector. For simplicity, we assume that this system is  , randomized Kaczmarz repeatedly performs the following steps:
, randomized Kaczmarz repeatedly performs the following steps: . Here, and going forward,
. Here, and going forward,  denotes the
denotes the  is the
is the  .
. and compute their norms
and compute their norms  . Then, sampling can be done using any algorithm for sampling from a weighted list of items. But what if
. Then, sampling can be done using any algorithm for sampling from a weighted list of items. But what if ![\[\norm{a_i}^2 \le B \quad \text{for each } i = 1,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b7b15a8b20d62e80eaffc7132bb0a59f_l3.png "Rendered by QuickLaTeX.com")
![[-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7e5005e1a5c93f262ad8df6aaa18b0bc_l3.png "Rendered by QuickLaTeX.com") , then (1) holds with
, then (1) holds with  . To sample a row
. To sample a row  , we perform the following the rejection sampling procedure:
, we perform the following the rejection sampling procedure:
 uniformly at random (i.e.,
uniformly at random (i.e.,  is equally likely to be any row index between
is equally likely to be any row index between  , accept and set
, accept and set  . With the remaining probability
. With the remaining probability  , reject and return to step 1.
, reject and return to step 1. . Our goal is to choose a random index
. Our goal is to choose a random index  , i.e.,
, i.e.,  . We will call
. We will call  the target distribution. (Note that we do not require the weights
the target distribution. (Note that we do not require the weights  .)
.) , i.e., we can efficiently generate random
, i.e., we can efficiently generate random  . Further, suppose that the proposal distribution dominates the target distribution in the sense that
. Further, suppose that the proposal distribution dominates the target distribution in the sense that ![\[w_i \le \rho_i \quad \text{for each } i=1,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a0d27cac7ee81dcf9ce84566471fa613_l3.png "Rendered by QuickLaTeX.com")
 , accept and set
, accept and set  and
and  for all
for all  , after which the probability of accepting
, after which the probability of accepting  . Therefore, the probability of outputting
. Therefore, the probability of outputting ![\[\prob \{\text{$i$ accepted this loop}\} = \frac{\rho_i}{\sum_{j=1}^n \rho_j} \cdot \frac{w_i}{\rho_i} = \frac{w_i}{\sum_{j=1}^n \rho_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-008a86385c50eb3900eb45369c84bfd3_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{\text{any accepted this loop}\} = \sum_{i=1}^n \prob \{\text{$i$ accepted this loop}\} = \frac{\sum_{i=1}^n w_i}{\sum_{i=1}^n \rho_i}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cb40d71ca9735132882dbcda9dc34464_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{\text{$i$ accepted this loop} \mid \text{any accepted this loop}\} = \frac{\prob \{\text{$i$ accepted this loop}\}}{\prob \{\text{any accepted this loop}\}} = \frac{w_i}{\sum_{j=1}^n w_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-367c6d93b8ba796a67f15ee16b773630_l3.png "Rendered by QuickLaTeX.com")
 , the ratio of the total mass of the target
, the ratio of the total mass of the target  . Thus, rejection sampling will have a high acceptance rate if
. Thus, rejection sampling will have a high acceptance rate if  and a low acceptance rate if
and a low acceptance rate if  . The conclusion for practice is that rejection sampling is only computationally efficient if one has access to a good proposal distribution
. The conclusion for practice is that rejection sampling is only computationally efficient if one has access to a good proposal distribution  .
. , but this not all that rejection sampling can do. Indeed, one can also use rejection sampling for sampling a real-valued parameters
, but this not all that rejection sampling can do. Indeed, one can also use rejection sampling for sampling a real-valued parameters  or a multivariate parameter
or a multivariate parameter  from a given (unnormalized) probability density function (or, if one likes, an unnormalized probability measure).
from a given (unnormalized) probability density function (or, if one likes, an unnormalized probability measure). and
and  are necessary to define the sampling probabilities
are necessary to define the sampling probabilities  and
and  , as computing
, as computing  denote the initial matrix and
denote the initial matrix and  denote a trivial initial approximation. For
denote a trivial initial approximation. For  , RPCholesky performs the following steps:
, RPCholesky performs the following steps: with probability
with probability  .
. . Here,
. Here,  denotes the
denotes the  .
. .
. . With an optimized implementation, RPCholesky requires only
. With an optimized implementation, RPCholesky requires only  operations and evaluates just
operations and evaluates just 
 denote the first
denote the first  produced by
produced by ![\[\hat{A}^{(j)} = A(:,S_j) A(S_j,S_j)^{-1}A(S_j,:). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c5a0be1ad5b7cafd120e5abc7d960cdb_l3.png "Rendered by QuickLaTeX.com")
![\[A^{(j)} = A - \hat{A}^{(j)} = A - A(:,S_j) A(S_j,S_j)^{-1}A(S_j,:).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-53cba1b4310799b43671c408bfae7b77_l3.png "Rendered by QuickLaTeX.com")
![\[A^{(j)}_{ii} = A_{ii} - A(i,S_j) A(S_j,S_j)^{-1} A(S_j,i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8191f0c1bf950de4259a6e2a5c0868ab_l3.png "Rendered by QuickLaTeX.com")
 is cheap, just requiring some arithmetic involving matrices and vectors of size
is cheap, just requiring some arithmetic involving matrices and vectors of size  , but it is expensive to evaluate all of the
, but it is expensive to evaluate all of the  . (One may verify that, as required,
. (One may verify that, as required,  for all
for all  , sample random indices
, sample random indices  using rejection sampling with proposal distribution
using rejection sampling with proposal distribution  are evaluated on an as-needed basis using (2).
are evaluated on an as-needed basis using (2). and
and  . The first matrix
. The first matrix  to its
to its  . The other matrix
. The other matrix  . Let’s see how long it takes to compute their eigenvalue decompositions in MATLAB:
. Let’s see how long it takes to compute their eigenvalue decompositions in MATLAB: longer to compute the eigenvalues of the non-Hermitian matrix
longer to compute the eigenvalues of the non-Hermitian matrix  of eigenvectors for a Hermitian matrix
of eigenvectors for a Hermitian matrix  is a
is a  .
. . The matrix
. The matrix  of eigenvectors for a normal matrix
of eigenvectors for a normal matrix  is also unitary,
is also unitary,  . An important class of normal matrices are unitary matrices themselves. A unitary matrix
. An important class of normal matrices are unitary matrices themselves. A unitary matrix  .
.![\[C = H + iS, \quad H = \frac{C+C^*}{2}, \quad S = \frac{C-C^*}{2i}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6435c1243a04b1a05cbc51277bd9d254_l3.png "Rendered by QuickLaTeX.com")
 and
and  . We know from matrix theory that commuting Hermitian matrices are
. We know from matrix theory that commuting Hermitian matrices are  and
and  for diagonal matrices
for diagonal matrices  and
and  . Thus, given access to such
. Thus, given access to such ![\[C = Q(D_H+iD_S)Q^*.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cbafa3b686b90d2e7c4fb1371e751c29_l3.png "Rendered by QuickLaTeX.com")
 for this matrix has a repeated eigenvalue. Thus,
for this matrix has a repeated eigenvalue. Thus,  of the Hermitian and skew-Hermitian parts of
of the Hermitian and skew-Hermitian parts of  and
and  .
. unitary matrix, RandDiag takes 0.4 seconds, just as fast as the Hermitian eigendecomposition did.
unitary matrix, RandDiag takes 0.4 seconds, just as fast as the Hermitian eigendecomposition did. , a matrix
, a matrix  will also diagonalize
will also diagonalize  . But, for any specific choice of
. But, for any specific choice of  , there is a possibility of failure. To avoid this possibility, we can just pick
, there is a possibility of failure. To avoid this possibility, we can just pick  and
and  at random. It’s really as simple as that.
at random. It’s really as simple as that. with
with  , its (
, its ( , where
, where  is a
is a  is
is  test matrix. It takes about 2.5 seconds to run.
test matrix. It takes about 2.5 seconds to run. is the
is the  is also small:
is also small: , computing its (upper triangular)
, computing its (upper triangular)  , and setting
, and setting  . Cholesky QR is very fast, about
. Cholesky QR is very fast, about  faster than Householder QR for this example:
faster than Householder QR for this example: , about ten million times larger than for Householder QR!:
, about ten million times larger than for Householder QR!: . The condition number of
. The condition number of  , which is at the root of Cholesky QR’s loss of accuracy. Thus, Cholesky QR is only appropriate for matrices that are well-conditioned, having a small condition number
, which is at the root of Cholesky QR’s loss of accuracy. Thus, Cholesky QR is only appropriate for matrices that are well-conditioned, having a small condition number  , say
, say  .
. that
that  is well-conditioned. Then, since
is well-conditioned. Then, since  ; see
; see  . This step compresses the very tall matrix
. This step compresses the very tall matrix  to the much shorter matrix
to the much shorter matrix  .
. using Householder QR. Since the matrix
using Householder QR. Since the matrix  .
. . Observe that
. Observe that  , as desired.
, as desired. faster in our experiment:
faster in our experiment: