This post is about randomized algorithms for problems in computational science and a powerful set of tools, known as concentration inequalities, which can be used to analyze why they work. I’ve discussed why randomization can help in solving computational problems in a previous post; this post continues this discussion by presenting an example of a computational problem where, somewhat surprisingly, a randomized algorithm proves effective. We shall then use concentration inequalities to analyze why this method works.
Triangle Counting
Let’s begin our discussion of concentration inequalities by means of an extended example. Consider the following question: How many triangles are there in the Facebook network? That is, how many trios of people are there who are all mutual friends? While seemingly silly at first sight, this is actually a natural and meaningful question about the structure of the Facebook social network and is related to similar questions such as “How likely are two friends of a person to also be friends with each other?”
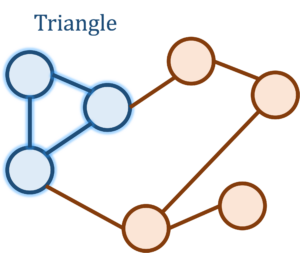
If there are  people on the Facebook graph, then the natural algorithm of iterating over all
people on the Facebook graph, then the natural algorithm of iterating over all  triplets and checking whether they form a triangle is far too computationally costly for the billions of Facebook accounts. Somehow, we want to do much faster than this, and to achieve this speed we would be willing to settle for an estimate of the triangle count up to some error.
triplets and checking whether they form a triangle is far too computationally costly for the billions of Facebook accounts. Somehow, we want to do much faster than this, and to achieve this speed we would be willing to settle for an estimate of the triangle count up to some error.
There are many approaches to this problem, but let’s describe a particularly surprising algorithm. Let  be an
be an  matrix where the
matrix where the  th entry of is
th entry of is  if users
if users  and
and  are friends and
are friends and  otherwise1All of the diagonal entries of are set to zero.; this matrix is called the adjacency matrix of the Facebook graph. A fact from graph theory is that the th entry of the cube
otherwise1All of the diagonal entries of are set to zero.; this matrix is called the adjacency matrix of the Facebook graph. A fact from graph theory is that the th entry of the cube  of the matrix counts the number of paths from user to user of length three.2By a path of length three, we mean a sequence of users
of the matrix counts the number of paths from user to user of length three.2By a path of length three, we mean a sequence of users  where and
where and  , and
, and  , and and are all friends. In particular, the
, and and are all friends. In particular, the  th entry of denotes the number of paths from to itself of length
th entry of denotes the number of paths from to itself of length  , which is twice the number of triangles incident on . (The paths
, which is twice the number of triangles incident on . (The paths  and
and  are both counted as paths of length 3 for a triangle consisting of , , and .) Therefore, the trace of , equal to the sum of its diagonal entries, is six times the number of triangles: The th entry of is twice the number of triangles incident on and each triangle
are both counted as paths of length 3 for a triangle consisting of , , and .) Therefore, the trace of , equal to the sum of its diagonal entries, is six times the number of triangles: The th entry of is twice the number of triangles incident on and each triangle  is counted thrice in the th,
is counted thrice in the th,  th, and
th, and  th entries of . In summary, we have
th entries of . In summary, we have

Therefore, the triangle counting problem is equivalent to computing the trace of . Unfortunately, the problem of computing is, in general, very computationally costly. Therefore, we seek ways of estimating the trace of a matrix without forming it.
Randomized Trace Estimation
Motivated by the triangle counting problem from the previous section, we consider the problem of estimating the trace of a matrix  . We assume that we only have access to the matrix through matrix–vector products; that is, we can efficiently compute
. We assume that we only have access to the matrix through matrix–vector products; that is, we can efficiently compute  for a vector
for a vector  . For instance, in the previous example, the Facebook graph has many fewer friend relations (edges)
. For instance, in the previous example, the Facebook graph has many fewer friend relations (edges)  than the maximum possible amount of
than the maximum possible amount of  . Therefore, the matrix is sparse; in particular, matrix–vector multiplications with can be computed in around operations. To compute matrix–vector products with
. Therefore, the matrix is sparse; in particular, matrix–vector multiplications with can be computed in around operations. To compute matrix–vector products with  , we simply compute matrix–vector products with three times,
, we simply compute matrix–vector products with three times,  .
.
Here’s a very nifty idea to estimate the trace of using only matrix–vector products, originally due to Didier A. Girard and Michael F. Hutchinson. Choose to be a random vector whose entries are independent  -values, where each value
-values, where each value  and
and  occurs with equal
occurs with equal  probability. Then if one forms the expression
probability. Then if one forms the expression  . Since the entries of
. Since the entries of  and
and  are independent, the expectation of
are independent, the expectation of  is for
is for  and for
and for  . Consequently, by linearity of expectation, the expected value of
. Consequently, by linearity of expectation, the expected value of  is
is
![\begin{equation*} \mathbb{E} \, x^\top M x = \sum_{i,j=1}^n M_{ij} \mathbb{E} [x_ix_j] = \sum_{i = 1}^n M_{ii} = \operatorname{tr}(M). \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ffecd976e434817c2637bb1e4987723e_l3.png "Rendered by QuickLaTeX.com")
The average value of is equal to the trace of ! In the language of statistics, we might say that is an unbiased estimator for  . Thus, the efficiently computable quantity can serve as a (crude) estimate for .
. Thus, the efficiently computable quantity can serve as a (crude) estimate for .
While the expectation of  equals , any random realization of can deviate from by a non-neligible amount. Thus, to reduce the variability of the estimator , it is appropriate to take an average of multiple copies of this random estimate. Specifically, we draw random vectors with independent random entries
equals , any random realization of can deviate from by a non-neligible amount. Thus, to reduce the variability of the estimator , it is appropriate to take an average of multiple copies of this random estimate. Specifically, we draw random vectors with independent random entries  and compute the averaged trace estimator
and compute the averaged trace estimator
(1) 
The -sample trace estimator  remains an unbiased estimator for ,
remains an unbiased estimator for ,  , but with reduced variability. Quantitatively, the variance of is times smaller than the single-sample estimator :
, but with reduced variability. Quantitatively, the variance of is times smaller than the single-sample estimator :
(2) 
The Girard–Hutchinson trace estimator gives a natural way of estimating the trace of the matrix , a task which might otherwise be hard without randomness.3To illustrate what randomness is buying us here, it might be instructive to think about how one might try to estimate the trace of via matrix–vector products without the help of randomness. For the trace estimator to be a useful tool, an important question remains: How many samples are needed to compute to a given accuracy? Concentration inequalities answer questions of this nature.
Concentration Inequalities
A concentration inequality provides a bound on the probability a random quantity is significantly larger or smaller than its typical value. Concentration inequalities are useful because they allow us to prove statements like “With at least 99% probability, the randomized trace estimator with 100 samples produces an approximation of the trace which is accurate up to error no larger than  .” In other words, concentration inequalities can provide quantitative estimates of the likely size of the error when a randomized algorithm is executed.
.” In other words, concentration inequalities can provide quantitative estimates of the likely size of the error when a randomized algorithm is executed.
In this section, we shall introduce a handful of useful concentration inequalities, which we will apply to the randomized trace estimator in the next section. We’ll then discuss how these and other concentration inequalities can be derived in the following section.
Markov’s Inequality
Markov’s inequality is the most fundamental concentration inequality. When used directly, it is a blunt instrument, requiring little insight to use and producing a crude but sometimes useful estimate. However, as we shall see later, all of the sophisticated concentration inequalities that will follow in this post can be derived from a careful use of Markov’s inequality.
The wide utility of Markov’s inequality is a consequence of the minimal assumptions needed for its use. Let  be any nonnegative random variable. Markov’s inequality states that the probability that exceeds a level
be any nonnegative random variable. Markov’s inequality states that the probability that exceeds a level  is bounded by the expected value of over
is bounded by the expected value of over  . In equations, we have
. In equations, we have
(3) 
We stress the fact that we make no assumptions on how the random quantity is generated other than that is nonnegative.
As a short example of Markov’s inequality, suppose we have a randomized algorithm which takes one second on average to run. Markov’s inequality then shows that the probability the algorithm takes more than 100 seconds to run is at most  . This small example shows both the power and the limitation of Markov’s inequality. On the negative side, our analysis suggests that we might have to wait as much as 100 times the average runtime for the algorithm to complete running with 99% probability; this large huge multiple of 100 seems quite pessimistic. On the other hand, we needed no information whatsoever about how the algorithm works to do this analysis. In general, Markov’s inequality cannot be improved without more assumptions on the random variable .4For instance, imagine an algorithm which 99% of the time completes instantly and 1% of the time takes 100 seconds. This algorithm does have an average runtime of 1 second, but the conclusion of Markov’s inequality that the runtime of the algorithm can be as much as 100 times the average runtime with 1% probability is true.
. This small example shows both the power and the limitation of Markov’s inequality. On the negative side, our analysis suggests that we might have to wait as much as 100 times the average runtime for the algorithm to complete running with 99% probability; this large huge multiple of 100 seems quite pessimistic. On the other hand, we needed no information whatsoever about how the algorithm works to do this analysis. In general, Markov’s inequality cannot be improved without more assumptions on the random variable .4For instance, imagine an algorithm which 99% of the time completes instantly and 1% of the time takes 100 seconds. This algorithm does have an average runtime of 1 second, but the conclusion of Markov’s inequality that the runtime of the algorithm can be as much as 100 times the average runtime with 1% probability is true.
Chebyshev’s Inequality and Averages
The variance of a random variable describes the expected size of a random variable’s deviation from its expected value. As such, we would expect that the variance should provide a bound on the probability a random variable is far from its expectation. This intuition indeed is correct and is manifested by Chebyshev’s inequality. Let be a random variable (with finite expected value) and . Chebyshev’s inequality states that the probability that deviates from its expected value by more than is at most  :
:
(4) 
Chebyshev’s inequality is frequently applied to sums or averages of independent random quantities. Suppose  are independent and identically distributed random variables with mean
are independent and identically distributed random variables with mean  and variance
and variance  and let
and let  denote the average
denote the average

Since the random variables are independent,5In fact, this calculation works if are only pairwise independent or even pairwise uncorrelated. For algorithmic applications, this means that don’t have to be fully independent of each other; we just need any pair of them to be uncorrelated. This allows many randomized algorithms to be “derandomized“, reducing the amount of “true” randomness needed to execute an algorithm. the properties of variance entail that

where we use the fact that  . Therefore, by Chebyshev’s inequality,
. Therefore, by Chebyshev’s inequality,
(5) 
Suppose we want to estimate the mean by up to error  and are willing to tolerate a failure probability of
and are willing to tolerate a failure probability of  . Then setting the right-hand side of (5) to , Chebyshev’s inequality suggests that we need at most
. Then setting the right-hand side of (5) to , Chebyshev’s inequality suggests that we need at most
(6) 
samples to achieve this goal.
Exponential Concentration: Hoeffding and Bernstein
How happy should we be with the result (6) of applying Chebyshev’s inequality the average ? The central limit theorem suggests that should be approximately normally distributed with mean and variance  . Normal random variables have an exponentially small probability of being more than a few standard deviations above their mean, so it is natural to expect this should be true of as well. Specifically, we expect a bound roughly like
. Normal random variables have an exponentially small probability of being more than a few standard deviations above their mean, so it is natural to expect this should be true of as well. Specifically, we expect a bound roughly like
(7) 
Unfortunately, we don’t have a general result quite this nice without additional assumptions, but there are a diverse array of exponential concentration inequalities available which are quite useful in analyzing sums (or averages) of independent random variables that appear in applications.
Hoeffding’s inequality is one such bound. Let be independent (but not necessarily identically distributed) random variables and consider the average  . Hoeffding’s inequality makes the assumption that the summands are bounded, say within an interval
. Hoeffding’s inequality makes the assumption that the summands are bounded, say within an interval ![[a,b]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-24633e5f5ecac15cd8a4843bd4257f84_l3.png "Rendered by QuickLaTeX.com") .6There are also more general versions of Hoeffding’s inequality where the bound on each random variable is different. Hoeffding’s inequality then states that
.6There are also more general versions of Hoeffding’s inequality where the bound on each random variable is different. Hoeffding’s inequality then states that
(8) 
Hoeffding’s inequality is quite similar to the ideal concentration result (7) except with the variance  replaced by the potentially much larger quantity7Note that is always smaller than or equal to
replaced by the potentially much larger quantity7Note that is always smaller than or equal to  . .
. .
Bernstein’s inequality fixes this deficit in Hoeffding’s inequality at a small cost. Now, instead of assuming are bounded within the interval , we make the alternate boundedness assumption  for every
for every  . We continue to denote so that if are identically distributed, denotes the variance of each of . Bernstein’s inequality states that
. We continue to denote so that if are identically distributed, denotes the variance of each of . Bernstein’s inequality states that
(9) 
For small values of , Bernstein’s inequality yields exactly the kind of concentration that we would hope for from our central limit theorem heuristic (7). However, for large values of , we have

which is exponentially small in rather than  . We conclude that Bernstein’s inequality provides sharper bounds then Hoeffding’s inequality for smaller values of but weaker bounds for larger values of .
. We conclude that Bernstein’s inequality provides sharper bounds then Hoeffding’s inequality for smaller values of but weaker bounds for larger values of .
Chebyshev vs. Hoeffding vs. Bernstein
Let’s return to the situation where we seek to estimate the mean of independent and identically distributed random variables each with variance by using the averaged value . Our goal is to bound how many samples we need to estimate up to error ,  , except with failure probability at most . Using Chebyshev’s inequality, we showed that (see (7))
, except with failure probability at most . Using Chebyshev’s inequality, we showed that (see (7))

Now, let’s try using Hoeffding’s inequality. Suppose that are bounded in the interval . Then Hoeffding’s inequality (8) shows that

Bernstein’s inequality states that if lie in the interval ![[\mu-B,\mu+B]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f7ce61b66273f19f68ecee5ab82c228d_l3.png "Rendered by QuickLaTeX.com") for every
for every  , then
, then
(10) 
Hoeffding’s and Bernstein’s inequality show that we need roughly proportional to  samples are needed rather than proportional to
samples are needed rather than proportional to  . The fact that we need proportional to
. The fact that we need proportional to  samples to achieve error is a consequence of the central limit theorem and is something we would not be able to improve with any concentration inequality. What exponential concentration inequalities allow us to do is to improve the dependence on the failure probability from proportional to
samples to achieve error is a consequence of the central limit theorem and is something we would not be able to improve with any concentration inequality. What exponential concentration inequalities allow us to do is to improve the dependence on the failure probability from proportional to  to
to  , which is a huge improvement.
, which is a huge improvement.
Hoeffding’s and Bernstein’s inequalities both have a small drawback. For Hoeffding’s inequality, the constant of proportionality is rather than the true variance of the summands. Bernstein’s inequality gives us the “correct” constant of proportionality but adds a second term proportional to  ; for small values of , this term is dominated by the term proportional to but the second term can be relevant for larger values of .
; for small values of , this term is dominated by the term proportional to but the second term can be relevant for larger values of .
There are a panoply of additional concentration inequalities than the few we’ve mentioned. We give a selected overview in the following optional section.
Analysis of Randomized Trace Estimation
Let us apply some of the concentration inequalities we introduced in last section to analyze the randomized trace estimator. Our goal is not to provide the best possible analysis of the trace estimator,8More precise estimation for trace estimation applied to positive semidefinite matrices was developed by Gratton and Titley-Peloquin; see Theorem 4.5 of the following survey. but to demonstrate how the general concentration inequalities we’ve developed can be useful “out of the box” in analyzing algorithms.
In order to apply Chebyshev’s and Berstein’s inequalities, we shall need to compute or bound the variance of the single-sample trace estimtor , where is a random vector of independent -values. This is a straightforward task using properties of the variance:

Here,  is the covariance and
is the covariance and  is the matrix Frobenius norm. Chebyshev’s inequality (5), then gives
is the matrix Frobenius norm. Chebyshev’s inequality (5), then gives

Let’s now try applying an exponential concentration inequality. We shall use Bernstein’s inequality, for which we need to bound  . By the Courant–Fischer minimax principle, we know that is between
. By the Courant–Fischer minimax principle, we know that is between  and
and  where
where  and
and  are the smallest and largest eigenvalues of and
are the smallest and largest eigenvalues of and  is the Euclidean norm of the vector . Since all the entries of have absolute value , we have
is the Euclidean norm of the vector . Since all the entries of have absolute value , we have  so is between
so is between  and
and  . Since the trace equals the sum of the eigenvalues of , is also between and . Therefore,
. Since the trace equals the sum of the eigenvalues of , is also between and . Therefore,

where  denotes the matrix spectral norm. Therefore, by Bernstein’s inequality (9), we have
denotes the matrix spectral norm. Therefore, by Bernstein’s inequality (9), we have

In particular, (10) shows that

samples suffice to estimate to error with failure probability at most . Concentration inequalities easily furnish estimates for the number of samples needed for the randomized trace estimator.
We have now accomplished our main goal of using concentration inequalities to analyze the randomized trace estimator, which in turn can be used to solve the triangle counting problem. We leave some additional comments on trace estimation and triangle counting in the following bonus section.
 and
and  . Since we often don’t know good bounds for and
. Since we often don’t know good bounds for and  , one should really use the trace estimator together with an a posteriori error estimates for the trace estimator, which provide a confidence interval for the trace rather than a point estimate; see sections 4.5 and 4.6 in this survey for details.
, one should really use the trace estimator together with an a posteriori error estimates for the trace estimator, which provide a confidence interval for the trace rather than a point estimate; see sections 4.5 and 4.6 in this survey for details.
One can improve on the Girard–Hutchinson trace estimator by using a variance reduction technique. One such variance reduction technique was recently proposed under the name Hutch++, extending ideas by Arjun Singh Gambhir, Andreas Stathopoulos, and Kostas Orginos and Lin Lin. In effect, these techniques improve the number of samples needed to estimate the trace of a positive semidefinite matrix to relative error to proportional to  down from .
down from .
Several algorithms have been proposed for triangle counting, many of them randomized. This survey gives a comparison of different methods for the triangle counting problem, and also describes more motivation and applications for the problem.
Deriving Concentration Inequalities
Having introduced concentration inequalities and applied them to the randomized trace estimator, we now turn to the question of how to derive concentration inequalities. Learning how to derive concentration inequalities is more than a matter of mathematical completeness since one can often obtain better results by “hand-crafting” a concentration inequality for a particular application rather than applying a known concentration inequality. (Though standard concentration inequalities like Hoeffding’s and Bernstein’s often give perfectly adequate answers with much less work.)
Markov’s Inequality
At the most fundamental level, concentration inequalities require us to bound a probability by an expectation. In achieving this goal, we shall make a simple observation: The probability that is larger than or equal to is the expectation of a random variable  .9More generally, the probability of an event can be written as an expectation of the indicator random variable of that event. Here,
.9More generally, the probability of an event can be written as an expectation of the indicator random variable of that event. Here,  is an indicator function which outputs one if its input is larger than or equal to and zero otherwise.
is an indicator function which outputs one if its input is larger than or equal to and zero otherwise.
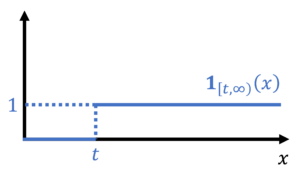
As promised, the probability is larger than is the expectation of :
(11) ![\begin{equation*} \mathbb{P}\{X \ge t \} = \mathbb{E}[\mathbf{1}_{[t,\infty)}(X)]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-231e1c7d5bf5d927c1b38eaa2c1bb4d8_l3.png "Rendered by QuickLaTeX.com")
We can now obtain bounds on the probability that  by bounding its corresponding indicator function. In particular, we have the inequality
by bounding its corresponding indicator function. In particular, we have the inequality
(12) 
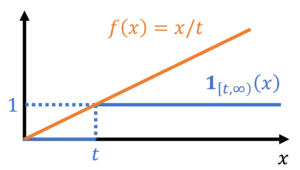
Since is nonnegative, combining equations (11) and (12) gives Markov’s inequality:
![\begin{equation*} \mathbb{P}\{ X \ge t \} = \mathbb{E}[\mathbf{1}_{[t,\infty)}(X)] \le \mathbb{E} \left[ \frac{X}{t} \right] = \frac{\mathbb{E} X}{t}. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7281716f886c12047b029b94dc4cab7b_l3.png "Rendered by QuickLaTeX.com")
Chebyshev’s Inequality
Before we get to Chebyshev’s inequality proper, let’s think about how we can push Markov’s inequality further. Suppose we find a bound on the indicator function of the form
(13) 
A bound of this form immediately to bounds on  by (11). To obtain sharp and useful bounds on
by (11). To obtain sharp and useful bounds on  we seek bounding functions
we seek bounding functions  in (13) with three properties:
in (13) with three properties:
- For
 ,
,  should be close to zero,
should be close to zero, - For
 , should be close to one, and
, should be close to one, and - We need
 to be easily computable or boundable.
to be easily computable or boundable.
These three objectives are in tension with each other. To meet criterion 3, we must restrict our attention to pedestrian functions such as powers  or exponentials
or exponentials  for which we have hopes of computing or bounding for random variables we encounter in practical applications. But these candidate functions have the undesirable property that making the function smaller on
for which we have hopes of computing or bounding for random variables we encounter in practical applications. But these candidate functions have the undesirable property that making the function smaller on  (by increasing
(by increasing  ) to meet point 1 makes the function larger on
) to meet point 1 makes the function larger on  , detracting from our ability to achieve point 2. We shall eventually come up with a best-possible resolution to this dilemma by formulating this as an optimization problem to determine the best choice of the parameter
, detracting from our ability to achieve point 2. We shall eventually come up with a best-possible resolution to this dilemma by formulating this as an optimization problem to determine the best choice of the parameter  to obtain the best possible candidate function of the given form.
to obtain the best possible candidate function of the given form.
Before we get ahead of ourselves, let us use a specific choice for different than we used to prove Markov’s inequality. We readily verify that  satisfies the bound (13), and thus by (12),
satisfies the bound (13), and thus by (12),
(14) 
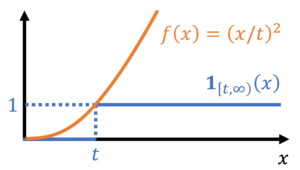
This inequality holds for any nonnegative random variable . In particular, now consider a random variable which we do not assume to be nonnegative. Then ‘s deviation from its expectation,  , is a nonnegative random variable. Thus applying (14) gives
, is a nonnegative random variable. Thus applying (14) gives

We have derived Chebyshev’s inequality! Alternatively, one can derive Chebyshev’s inequality by noting that  if, and only if,
if, and only if,  . Therefore, by Markov’s inequality,
. Therefore, by Markov’s inequality,

The Laplace Transform Method
We shall now realize the plan outlined earlier where we shall choose an optimal bounding function from the family of exponential functions  , where is a parameter which we shall optimize over. This method shall allow us to derive exponential concentration inequalities like Hoeffding’s and Bernstein’s. Note that the exponential function bounds the indicator function for all real numbers , so we shall no longer require the random variable to be nonnegative. Therefore, by (11),
, where is a parameter which we shall optimize over. This method shall allow us to derive exponential concentration inequalities like Hoeffding’s and Bernstein’s. Note that the exponential function bounds the indicator function for all real numbers , so we shall no longer require the random variable to be nonnegative. Therefore, by (11),
(15) 
The functions

are known as the moment generating function and cumulant generating function of the random variable .10These functions are so-named because they are the (exponential) generating functions of the (polynomial) moments  ,
,  , and the cumulants of . With these notations, (15) can be written
, and the cumulants of . With these notations, (15) can be written
(16) 
The moment generating function coincides with the Laplace transform  up to the sign of the parameter , so one name for this approach to deriving concentration inequalities is the Laplace transform method. (This method is also known as the Cramér–Chernoff method.)
up to the sign of the parameter , so one name for this approach to deriving concentration inequalities is the Laplace transform method. (This method is also known as the Cramér–Chernoff method.)
The cumulant generating function has an important property for deriving concentration inequalities for sums or averages of independent random variables: If are independent random variables, than the cumulant generating function is additive:11For proof, we compute  . Taking logarithms proves the additivity.
. Taking logarithms proves the additivity.
(17) 
Proving Hoeffding’s Inequality
For us to use the Laplace transform method, we need to either compute or bound the cumulant generating function. Since we are interested in general concentration inequalities which hold under minimal assumptions such as boundedness, we opt for the latter. Suppose  and consider the cumulant generating function of
and consider the cumulant generating function of  . Then one can show the cumulant generating function bound12The bound (18) is somewhat tricky to establish, but we can establish the same result with a larger constant than
. Then one can show the cumulant generating function bound12The bound (18) is somewhat tricky to establish, but we can establish the same result with a larger constant than  . We have
. We have  . Since the function
. Since the function  is convex, we have the bound
is convex, we have the bound  . Taking expectations, we have
. Taking expectations, we have  . One can show by comparing Taylor series that
. One can show by comparing Taylor series that  . Therefore, we have
. Therefore, we have  .
.
(18) 
Using the additivity of the cumulant generating function (17), we obtain the bound

Plugging this into the probability bound (16), we obtain the concentration bound
(19) 
We want to obtain the smallest possible upper bound on this probability, so it behooves us to pick the value of which makes the right-hand side of this inequality as small as possible. To do this, we differentiate the contents of the exponential and set to zero, obtaining

Plugging this value for into the bound (19) gives A bound for being larger than  :
:
(20) 
To get the bound on being smaller than  , we can apply a small trick. If we apply (20) to the summands
, we can apply a small trick. If we apply (20) to the summands  instead of , we obtain the bounds
instead of , we obtain the bounds
(21) 
We can now combine the upper tail bound (19) with the lower tail bound (21) to obtain a “symmetric” bound on the probability that  . The means of doing often this goes by the fancy name union bound, but the idea is very simple:
. The means of doing often this goes by the fancy name union bound, but the idea is very simple:

Thus, applying this union bound idea with the upper and lower tail bounds (20) and (21), we obtain Hoeffding’s inequality, exactly as it appeared above as (8):

Voilà! Hoeffding’s inequality has been proven! Bernstein’s inequality is proven essentially the same way except that, instead of (17), we have the cumulant generating function bound

for a random variable  with mean zero and satisfying the bound
with mean zero and satisfying the bound  .
.
Upshot: Randomness can be a very effective tool for solving computational problems, even those which seemingly no connection to probability like triangle counting. Concentration inequalities are a powerful tool for assessing how many samples are needed for an algorithm based on random sampling to work. Some of the most useful concentration inequalities are exponential concentration inequalities like Hoeffding and Bernstein, which show that an average of bounded random quantities are close to their average except with exponentially small probability.
 matrix
matrix  of length
of length  is as close to
is as close to  . That is, we seek to
. That is, we seek to 
 of
of  of
of  , our model predicts the output
, our model predicts the output  is approximately
is approximately  . The vector
. The vector  is as small as it could possibly be for all choices of coefficient vectors
is as small as it could possibly be for all choices of coefficient vectors 
 is invertible and the unique least squares solution to (1) is given by
is invertible and the unique least squares solution to (1) is given by  . We assume that
. We assume that  and solve (2) using a linear solver like
and solve (2) using a linear solver like  on a computer using a well-designed piece of software, one obtains an approximate solution
on a computer using a well-designed piece of software, one obtains an approximate solution  which is, after accounting for the accumulation of rounding errors, close to
which is, after accounting for the accumulation of rounding errors, close to  .
. of the largest and smallest
of the largest and smallest 
 corresponds to the fact we have roughly 16 decimal digits of accuracy in double precision floating point arithmetic. Thus, if the condition number of
corresponds to the fact we have roughly 16 decimal digits of accuracy in double precision floating point arithmetic. Thus, if the condition number of  , then we should expect around
, then we should expect around  digits of accuracy in our computed solution.
digits of accuracy in our computed solution. . We would hope that we can solve the least squares problem with an accuracy like the rule-of-thumb error bound (3) we had for linear systems of equations, namely a bound like
. We would hope that we can solve the least squares problem with an accuracy like the rule-of-thumb error bound (3) we had for linear systems of equations, namely a bound like  . But this is not the kind of accuracy we get for the least squares problem when we solve it using the normal equations. Instead, we get accuracy like
. But this is not the kind of accuracy we get for the least squares problem when we solve it using the normal equations. Instead, we get accuracy like
 , then the normal equations give us absolute nonsense for
, then the normal equations give us absolute nonsense for  times larger than
times larger than  . So even if we solve the least squares problem with QR factorization, we still get a squared condition number in our error bound, but this condition number squared is multiplied by the residual
. So even if we solve the least squares problem with QR factorization, we still get a squared condition number in our error bound, but this condition number squared is multiplied by the residual  , which is small if the least squares fit is good. The least squares solution is usually only interesting when the residual is small, thus justifying dropping it in the rule of thumb.
, which is small if the least squares fit is good. The least squares solution is usually only interesting when the residual is small, thus justifying dropping it in the rule of thumb.
 ” matrix in the QR factorization can be represented implicitly as a product of easy-to-compute-with Householder reflectors which is much more efficient when
” matrix in the QR factorization can be represented implicitly as a product of easy-to-compute-with Householder reflectors which is much more efficient when , etc.
, etc. denote the temperature of the material at point
denote the temperature of the material at point  . Laplace’s equation states that, at any point
. Laplace’s equation states that, at any point 

 .
. on the boundary to produce the electric potential
on the boundary to produce the electric potential  be the region of interest and
be the region of interest and  its boundary. Denote points
its boundary. Denote points  , with the length of
, with the length of  denoted
denoted  . The double layer potential satisfies
. The double layer potential satisfies
 denotes the directional derivative taken in the direction normal (perpendicular) to the surface at the point
denotes the directional derivative taken in the direction normal (perpendicular) to the surface at the point  . Note we choose a unit system for
. Note we choose a unit system for  which hides physical constants particular to the electrostatic context, since we are interested in applying this methodology to the Laplace Dirichlet problem in general (possibly non-electrostatic) applications.
which hides physical constants particular to the electrostatic context, since we are interested in applying this methodology to the Laplace Dirichlet problem in general (possibly non-electrostatic) applications. at points
at points 
 centered at
centered at 


 ,
,  , and
, and  in memory and multiplying it with
in memory and multiplying it with  .
. .
. equations for unknowns
equations for unknowns  . Specifically, our first equation can be rewritten as
. Specifically, our first equation can be rewritten as




 , we have that
, we have that

 from
from  but, as we did last section, let’s first consider the reverse. To compute
but, as we did last section, let’s first consider the reverse. To compute 


 times equation (8) to equation (7). Following last section, we now instead eliminate
times equation (8) to equation (7). Following last section, we now instead eliminate  from equation (8) using equation (7). To do this, we need to integrate equation (7) in order to cancel the integral in equation (8):
from equation (8) using equation (7). To do this, we need to integrate equation (7) in order to cancel the integral in equation (8):
 times this integrated equation to equation (8) yields
times this integrated equation to equation (8) yields


 using the
using the 
 for the
for the  for the normal vector to
for the normal vector to  for the
for the  for the
for the  is a so-called
is a so-called  . Therefore, (9) simplifies to
. Therefore, (9) simplifies to

 for points
for points 
 is some region in space,
is some region in space,  ,
,  is a nonzero constant. Such an integral equation is said to be of the
is a nonzero constant. Such an integral equation is said to be of the  ,
,  ,
,  , and
, and  . We say the kernel
. We say the kernel 
 .
. and
and  .
. ):
):
 denote the identity operator on functions: It takes as inputs function
denote the identity operator on functions: It takes as inputs function  denote the “integration against
denote the “integration against  operator”: It takes as input a function
operator”: It takes as input a function  .
.


 system of linear equations. One can use this as a numerical method to solve second-kind integral equations: First, we
system of linear equations. One can use this as a numerical method to solve second-kind integral equations: First, we  by a separable kernel of a modest rank
by a separable kernel of a modest rank  -decompositions, sparse component analysis, and the blind separation of sums of exponentials
-decompositions, sparse component analysis, and the blind separation of sums of exponentials![\[f(t) = a_0 \cos(\omega_0 t - \phi_0) + a_1 \cos(\omega_1 t - \phi_1) + a_2 \cos(\omega_2 t - \phi_2). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8e44bb98be117230aa4cd5148d841129_l3.png "Rendered by QuickLaTeX.com")
 ,
,  , and
, and  .
. at certain times
at certain times  which we assume are equally spaced
which we assume are equally spaced![\[t_j = j\Delta t \quad \textnormal{for} \quad j = 0,1,2,\ldots,n-1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-814820bd1ef46e2fee855eb68600d927_l3.png "Rendered by QuickLaTeX.com")
![\[f_j = f(t_j) \quad \textnormal{for} \quad j = 0,1,2,\ldots,n-1\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a0b9511cab842078a47a27836c5821e1_l3.png "Rendered by QuickLaTeX.com")
 , where
, where  it will be helpful to rewrite our formula (1) for
it will be helpful to rewrite our formula (1) for ![\[\cos \alpha = \frac{\mathrm{e}^{\mathrm{i} \alpha}+\mathrm{e}^{-\mathrm{i} \alpha}}{2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ffb0475e120f3b8c6340c432631c5962_l3.png "Rendered by QuickLaTeX.com")
![\[a_0 \cos(\omega_0 t - \phi_0) = d_0 \mathrm{e}^{\mathrm{i} \omega_0t} + d_1 \mathrm{e}^{-\mathrm{i} \omega_0t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1683f7679dca96f66c9221332d5e0bdb_l3.png "Rendered by QuickLaTeX.com")
 and
and  are complex coefficients
are complex coefficients  and
and  which contain the same information as the original parameters
which contain the same information as the original parameters  and
and  . Now notice that we are only interest in values
. Now notice that we are only interest in values  which are multiples of the spacing
which are multiples of the spacing  . Thus, our frequency component can be further rewritten as
. Thus, our frequency component can be further rewritten as![\[a_0 \cos(\omega_0 t_j - \phi_0) = d_0 z_0^j + d_1 z_1^j\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-269ef790089c772d8c424bd62aa1707e_l3.png "Rendered by QuickLaTeX.com")
 and
and  . Performing these reductions, our samples
. Performing these reductions, our samples  take the form
take the form![\[f_j = d_0 z_0^j + d_1 z_1^j + \cdots + d_5 z_5^j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a32d21e3f31302be06340549f7bf65b2_l3.png "Rendered by QuickLaTeX.com")
 and
and  in the relation (2) from a small number of measurements
in the relation (2) from a small number of measurements  . This can reveal patterns in the sequence which are less obvious when it is represented as given just as numbers. For instance, any seven columns of
. This can reveal patterns in the sequence which are less obvious when it is represented as given just as numbers. For instance, any seven columns of  which can be much larger than seven. In addition, as we will soon effectively exploit later, vectors in the nullspace of
which can be much larger than seven. In addition, as we will soon effectively exploit later, vectors in the nullspace of ![\[H = \begin{bmatrix} f_0 & f_1 & f_2 & \cdots & f_{(n-1)/2} \\f_1 & f_2 & f_3 & \cdots & f_{(n-1)/2+1} \\f_2 & f_3 & f_4 & \cdots & f_{(n-1)/2+2} \\\vdots & \vdots & \vdots & \ddots & \vdots \\f_{(n-1)/2} & f{(n-1)/2+1} & f{(n-1)/2+2} & \cdots & f_{n-1} \end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-42f02441b9724e66513f1e422c797113_l3.png "Rendered by QuickLaTeX.com")
 . Observe that the entry in position
. Observe that the entry in position  . (We use a
. (We use a  using (2):
using (2):![\[H_{ij} = f_{i+j} = \sum_{k=0}^5 d_k z_k^{i+j} = \sum_{k=0}^5 d_k z_k^i \cdot z_k^j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c40e19f060d1924406303ffb887b2153_l3.png "Rendered by QuickLaTeX.com")
 was just begging to be factorized as
was just begging to be factorized as  , which we did. Equation (4) almost looks like the formula for the product of two matrices with entries
, which we did. Equation (4) almost looks like the formula for the product of two matrices with entries  , so it makes sense to introduce the
, so it makes sense to introduce the  matrix
matrix  with entry
with entry  . This is a so-called
. This is a so-called ![\[V = \begin{bmatrix}z_0^0 & z_0^1 & z_0^2 & \cdots & z_0^{(n-1)/2} \\z_1^0 & z_1^1 & z_1^2 & \cdots & z_1^{(n-1)/2} \\\vdots & \vdots & \vdots & \ddots & \vdots \\z_5^0 & z_5^1 & z_5^2 & \cdots & z_5^{(n-1)/2}\end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a1d818b7fab7cb659d2a20c9f94bc743_l3.png "Rendered by QuickLaTeX.com")
 diagonal matrix
diagonal matrix  , the formula (4) for
, the formula (4) for  , the factor
, the factor ![\[H = V^\top D V. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2a7b807a19d51a8bd8d8de2e04aac3f3_l3.png "Rendered by QuickLaTeX.com")
 describing our sampled recording
describing our sampled recording  , determining all the frequencies would be very straightforward.
, determining all the frequencies would be very straightforward. of
of ![\[f_m = c_{k-1} f_{m-1} + \cdots + c_1f_{m-k+1}+ c_0 f_{m-k} \quad \textnormal{for every} \quad m = k,\,{k+1},\,{k+2},\cdots. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-62d879500458ed1476a089397147f5c4_l3.png "Rendered by QuickLaTeX.com")
 because there are six terms in the formula (2) for
because there are six terms in the formula (2) for  , such a recurrence will allow us to determine the parameters
, such a recurrence will allow us to determine the parameters  . A very good rule of thumb in applied mathematics is to always write down linear equations in matrix–vector notation in see how it looks. Doing this, we obtain
. A very good rule of thumb in applied mathematics is to always write down linear equations in matrix–vector notation in see how it looks. Doing this, we obtain![\[\begin{bmatrix} f_6 \\ f_7 \\ \vdots \\ f_{n-1} \end{bmatrix} = \underbrace{\begin{bmatrix} f_0 & f_1 & f_2 & f_3 & f_4 & f_5 \\ f_1 & f_2 & f_3 & f_4 & f_5 & f_6 \\ \vdots & \vdots & \vdots & \vdots & \vdots & \vdots \\ f_{n-7} & f_{n-6} & f_{n-5} & f_{n-4} & f_{n-3} & f_{n-2} \end{bmatrix}}_{=F}\begin{bmatrix} c_0 \\ c_1 \\ \vdots \\ c_5 \end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-aa5891d632cb53b7b006e08328f5d3a0_l3.png "Rendered by QuickLaTeX.com")
 . Unlike
. Unlike  going forward.
going forward. would also be fine for our purposes, but we assume
would also be fine for our purposes, but we assume ![\[f_{6 \, :\, n-1} = F c, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-206cb0e274493646eb686f2fc6b773df_l3.png "Rendered by QuickLaTeX.com")
 for the vector on the left-hand side of (7) and collected the recurrence coefficients
for the vector on the left-hand side of (7) and collected the recurrence coefficients  . For a typical system of linear equations like (8), we would predict the system to have no solution
. For a typical system of linear equations like (8), we would predict the system to have no solution  ), the system equations (8) has more equations than unknowns. Fortunately, we are not in the typical case. Despite the fact that we have more equations than unknowns, the linear equations (8) have a unique solution
), the system equations (8) has more equations than unknowns. Fortunately, we are not in the typical case. Despite the fact that we have more equations than unknowns, the linear equations (8) have a unique solution  In particular, the matrix on the right-hand side of this equation is guaranteed to be nonsingular under our assumptions. Using the Vandermonde decomposition, can you see why?
In particular, the matrix on the right-hand side of this equation is guaranteed to be nonsingular under our assumptions. Using the Vandermonde decomposition, can you see why? ; we seek to find all complex numbers
; we seek to find all complex numbers  for which this is a bonafide solution. Plugging this solution into the formula (6) for
for which this is a bonafide solution. Plugging this solution into the formula (6) for  gives
gives![\[z^6 = c_0 z^5 + c_1 z^4 + \cdots + c_6. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c36ec055ae832307db7d77cb25d469a_l3.png "Rendered by QuickLaTeX.com")
![\[\begin{bmatrix}f_0 \\ f_1 \\ \vdots \\ f_{n-1}\end{bmatrix} = \begin{bmatrix}1 & 1 & \cdots & 1 \\ z_0 & z_1 & \cdots & z_5 \\ \vdots & \vdots & \ddots & \vdots \\ z_0^{n-1} & z_1^{n-1} & \cdots & z_5^{n-1}\end{bmatrix} \begin{bmatrix}d_0 \\ d_1 \\ \vdots \\ d_{n-1}.\end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b1cdea36621ca6c0c1c791764d993fea_l3.png "Rendered by QuickLaTeX.com")
 . So far, the benefits of the matrix factorization perspective have yet to really reveal themselves.
. So far, the benefits of the matrix factorization perspective have yet to really reveal themselves. needed to describe the samples and the representation of the samples as a mixture of exponentials is uniquely determined only if the matrix
needed to describe the samples and the representation of the samples as a mixture of exponentials is uniquely determined only if the matrix  ,
,  , and
, and  . (Here, the superscript denotes an index rather than differentiation.) Each signal is a weighted mixture of three sources
. (Here, the superscript denotes an index rather than differentiation.) Each signal is a weighted mixture of three sources  ,
,  , and
, and  , each of which plays a musical chord of three notes (thus representable as a sum of cosines as in (1)). One can think of the sources of being produced three different musical instruments at different places in a room and the recordings
, each of which plays a musical chord of three notes (thus representable as a sum of cosines as in (1)). One can think of the sources of being produced three different musical instruments at different places in a room and the recordings  for each recording
for each recording  and form each collection of samples into a Hankel matrix
and form each collection of samples into a Hankel matrix![\[H^{(\ell)} = \begin{bmatrix} f_0^{(\ell)} & f_1^{(\ell)} & f_2^{(\ell)} & \cdots & f_{(n-1)/2}^{(\ell)} \\f_1^{(\ell)} & f_2^{(\ell)} & f_3^{(\ell)} & \cdots & f_{(n-1)/2+1}^{(\ell)} \\f_2^{(\ell)} & f_3^{(\ell)} & f_4^{(\ell)} & \cdots & f_{(n-1)/2+2}^{(\ell)} \\\vdots & \vdots & \vdots & \ddots & \vdots \\f_{(n-1)/2}^{(\ell)} & f_{(n-1)/2+1}^{(\ell)} & f_{(n-1)/2+2}^{(\ell)} & \cdots & f_{n-1}^{(\ell)} \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5c3bb6afd39df0eba56eaeee8c08a77f_l3.png "Rendered by QuickLaTeX.com")
 ,
,  , and
, and  as slices of a
as slices of a  . A tensor,
. A tensor,  array given by
array given by![\[\mathcal{H}(:,:,\ell) = H^{(\ell)} \quad \textnormal{for} \quad \ell=1,2,3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7a46aa5e41df12034dc3c63905a626ba_l3.png "Rendered by QuickLaTeX.com")
![\[H = V^\top DV = \tilde{V}^\top \tilde{D} \tilde{V}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a480b33a17f5c4bbe9f3e057a9350924_l3.png "Rendered by QuickLaTeX.com")
 can be uniquely determined from the measurements
can be uniquely determined from the measurements  rows,
rows,  is
is  ‘s are all uniquely determined.
‘s are all uniquely determined.  and perhaps
and perhaps  ,
,  , etc.
, etc. This matrix has rank-two but no rank-two confluent Vandermonde decomposition. The issue is that when extended to an infinite Hankel matrix
This matrix has rank-two but no rank-two confluent Vandermonde decomposition. The issue is that when extended to an infinite Hankel matrix  this (infinite!) matrix has a rank exceeding the size of the original Hankel matrix
this (infinite!) matrix has a rank exceeding the size of the original Hankel matrix  into a Hankel matrix, it can also be useful to form them into a
into a Hankel matrix, it can also be useful to form them into a ![\[T = \begin{bmatrix} f_{(n-1)/2} & f_{(n-1)/2+1} & f_{(n-1)/2+2} & \cdots & f_{n-1} \\\\f_{(n-1)/2-1} & f_{(n-1)/2} & f_{(n-1)/2+1} & \cdots & f_{n-2} \\\\f_{(n-1)/2-2} & f_{(n-1)/2-1} & f_{(n-1)/2} & \cdots & f_{n-3} \\\\\vdots & \vdots & \vdots & \ddots & \vdots \\\\ f_0 & f_1 & f_2 & \cdots & f_{(n-1)/2} \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a99cb184de9f3b17cda900a0997b483c_l3.png "Rendered by QuickLaTeX.com")
 has the appealing propery that the matrix–vector product
has the appealing propery that the matrix–vector product  computes a
computes a ![\[T = \operatorname{ReversedRows}(V^\top) \cdot DV.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-66be95d184f371b43d13547ce6eb5a81_l3.png "Rendered by QuickLaTeX.com")
![\[T = V^* D V,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ef493ca7d7b0327e179da261e9f32ddb_l3.png "Rendered by QuickLaTeX.com")
 which are
which are  where
where  .
. such that
such that  .
. will be real
will be real  , which is a special case of the generalized eigenvalue problem with
, which is a special case of the generalized eigenvalue problem with  .
. denotes a vector of generalized coordinates describing our system and
denotes a vector of generalized coordinates describing our system and  denotes
denotes  are given by the
are given by the  . If we choose
. If we choose  is a (static) equilibrium configuration for which
is a (static) equilibrium configuration for which  whenever
whenever  .
.

 , where
, where  .
. can be written (uniquely) as linear combinations of solutions of the form
can be written (uniquely) as linear combinations of solutions of the form  . For several reasons, this is a less-than-appealing way of reducing a generalized eigenvalue problem to a standard eigenvalue problem. A better way, appropriate when
. For several reasons, this is a less-than-appealing way of reducing a generalized eigenvalue problem to a standard eigenvalue problem. A better way, appropriate when  , which also possesses the same eigenvalues as
, which also possesses the same eigenvalues as  .
. compare to those of
compare to those of  and
and  , where
, where  is the
is the  .) For the remainder of this post, let
.) For the remainder of this post, let  and
and  denote the perturbations.
denote the perturbations. , then
, then  . That is,
. That is,  is an eigenvalue of the pair
is an eigenvalue of the pair  . Lack of symmetry is an ugly feature in a mathematical theory, so we seek to smooth it out. After thinking a little bit, notice that we can phrase the generalized eigenvalue condition symmetrically as
. Lack of symmetry is an ugly feature in a mathematical theory, so we seek to smooth it out. After thinking a little bit, notice that we can phrase the generalized eigenvalue condition symmetrically as  with the associated eigenvalue being given by
with the associated eigenvalue being given by  . This observation may seem trivial at first, but let us collect it for good measure.
. This observation may seem trivial at first, but let us collect it for good measure. .
. ? The case
? The case  , this expression still makes sense: we’ve found a vector
, this expression still makes sense: we’ve found a vector  but
but  . It makes sense to consider
. It makes sense to consider  ! Dividing by zero should justifiably make one squeemish, but it really is quite natural in the case to treat
! Dividing by zero should justifiably make one squeemish, but it really is quite natural in the case to treat  .
. . Then, any
. Then, any  can reasonably considered an eigenvalue of
can reasonably considered an eigenvalue of  . In such a case, all complex numbers are simultaneously eigenvalues of
. In such a case, all complex numbers are simultaneously eigenvalues of  is identically zero for all
is identically zero for all  .
. . Thus, it’s better not to think of
. Thus, it’s better not to think of  for
for 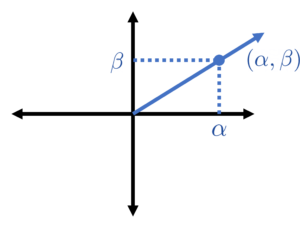
 and rearranging gives
and rearranging gives  —the eigenvalue
—the eigenvalue  , which is so important it is given a name: the
, which is so important it is given a name: the 
 are only determined up to a scaling factor, it shows we can take
are only determined up to a scaling factor, it shows we can take  and
and  . And by different scalings of the eigenvector
. And by different scalings of the eigenvector  and
and  by any positive factor we want. (This retroactively shows why it makes sense to only consider positive scalings of
by any positive factor we want. (This retroactively shows why it makes sense to only consider positive scalings of  . Thus, even though
. Thus, even though  lead to equivalent eigenvalues since
lead to equivalent eigenvalues since  ,
,  , this does not mean there exists
, this does not mean there exists  such that
such that  . Therefore, we only consider
. Therefore, we only consider  is so important that we give it a name: the
is so important that we give it a name: the  .
. is real, so we can pick a scaling in which both
is real, so we can pick a scaling in which both 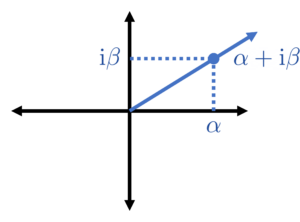
 is best thought of as a pair
is best thought of as a pair 
 .If we just went straight from one to the other, this reduction would appear like some crazy stroke of inspiration: why would I ever think to write down
.If we just went straight from one to the other, this reduction would appear like some crazy stroke of inspiration: why would I ever think to write down  , we have that
, we have that  : consequently,
: consequently,  is a small perturbation of
is a small perturbation of  is
is  as well and thus
as well and thus  . It is thus quite natural to assume the following definiteness condition:
. It is thus quite natural to assume the following definiteness condition: for all complex nonzero vectors
for all complex nonzero vectors  .
.  . However,
. However,  is not identically zero..
is not identically zero.. for all vectors
for all vectors  is just scaled by a positive factor by scaling the vector
is just scaled by a positive factor by scaling the vector  of a pair
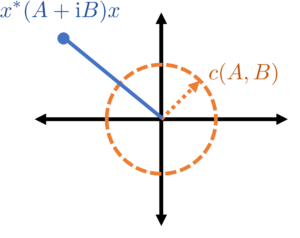
of a pair  over all complex unit vectors
over all complex unit vectors  .
.
 (in our case
(in our case  ), the set of complex numbers
), the set of complex numbers  for all unit vectors
for all unit vectors  .
. to zero.
to zero. for a definite matrix pair
for a definite matrix pair  .
. for unit vectors
for unit vectors 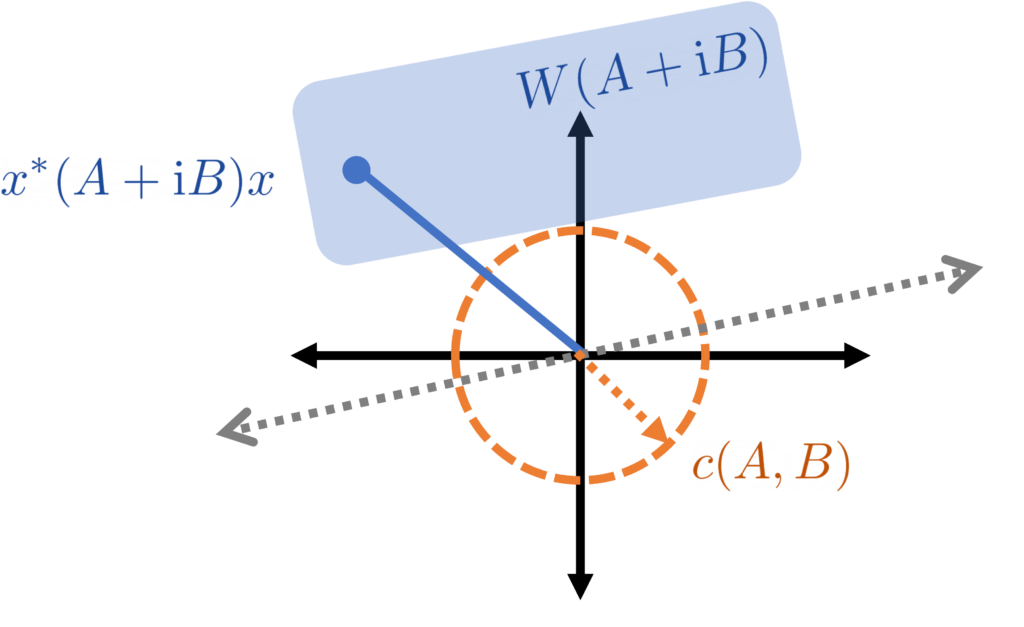
 radians). As such, to associate each ray a unique angle we need to pin down this indeterminacy in some fixed way. Moreover, this indeterminacy should play nice with the field of values
radians). As such, to associate each ray a unique angle we need to pin down this indeterminacy in some fixed way. Moreover, this indeterminacy should play nice with the field of values  of the perturbation. But in the last section, we say that each of these field of angles lies (strictly) on one half of the complex plane. Thus, we can find a ray
of the perturbation. But in the last section, we say that each of these field of angles lies (strictly) on one half of the complex plane. Thus, we can find a ray  for some real number
for some real number  . The argument is multi-valued since
. The argument is multi-valued since  is an argument for
is an argument for  as long as
as long as  . If
. If  an eigenangle.
an eigenangle. .
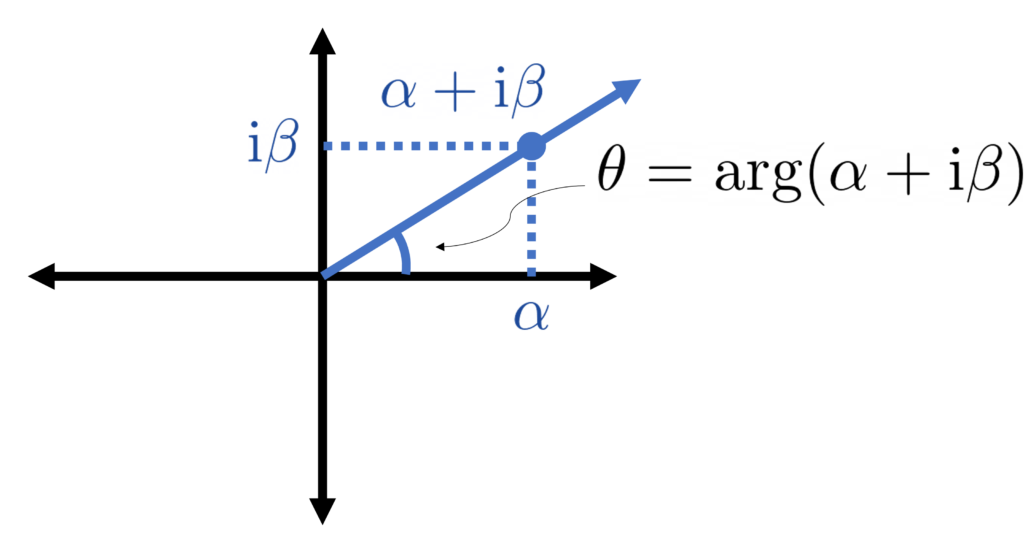
.


 can be written as the following minimax optimization problem
can be written as the following minimax optimization problem
 of dimension
of dimension  whereas the maximum is taken over all unit vectors
whereas the maximum is taken over all unit vectors 


 .
.![[0,\pi]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-049f2026ce1810bcad11310197f7804b_l3.png "Rendered by QuickLaTeX.com") , this means that the eigenvalues
, this means that the eigenvalues  are now in increasing order, in contrast to our convention from earlier in this section.
are now in increasing order, in contrast to our convention from earlier in this section.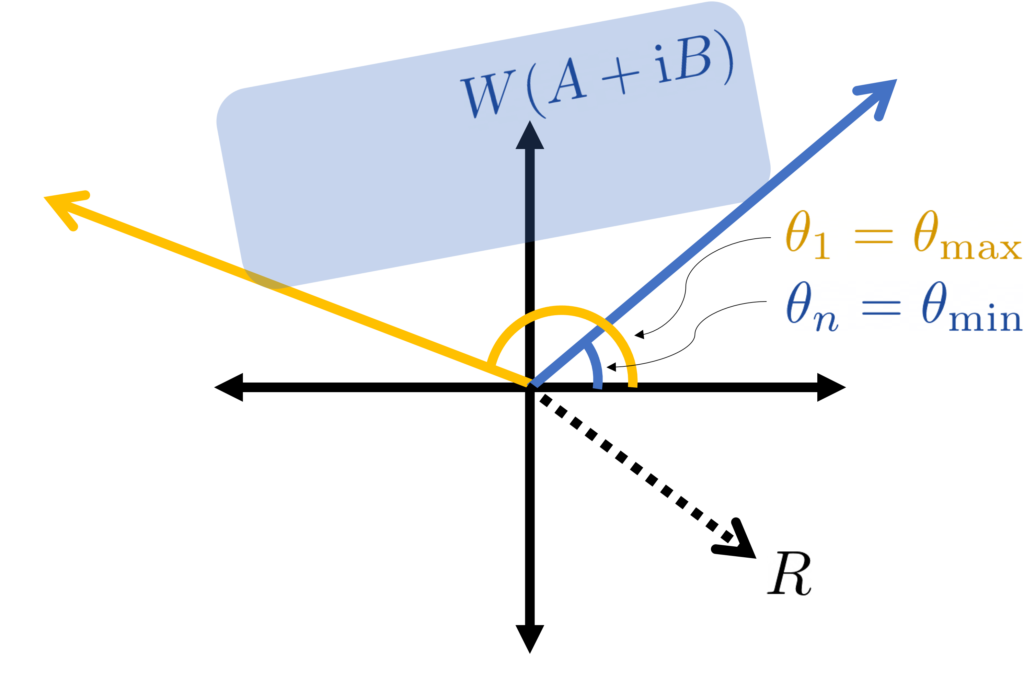
 by the distance between
by the distance between  . But the difference between these two quantities is just
. But the difference between these two quantities is just 
 be? If the vector
be? If the vector  from zero. This quantity has a name:
from zero. This quantity has a name: (in our case
(in our case  ) is
) is  .
. .
.
 by more familiar quantities. For instance, once can straightforwardly show the bound
by more familiar quantities. For instance, once can straightforwardly show the bound  , where
, where  denote the eigenangles of the perturbed pair
denote the eigenangles of the perturbed pair  and consider the
and consider the  be the subspace of dimension
be the subspace of dimension 
 . We’re truly toast if the perturbation is large enough to perturb
. We’re truly toast if the perturbation is large enough to perturb  to be equal to zero, so we should assume that
to be equal to zero, so we should assume that  .
.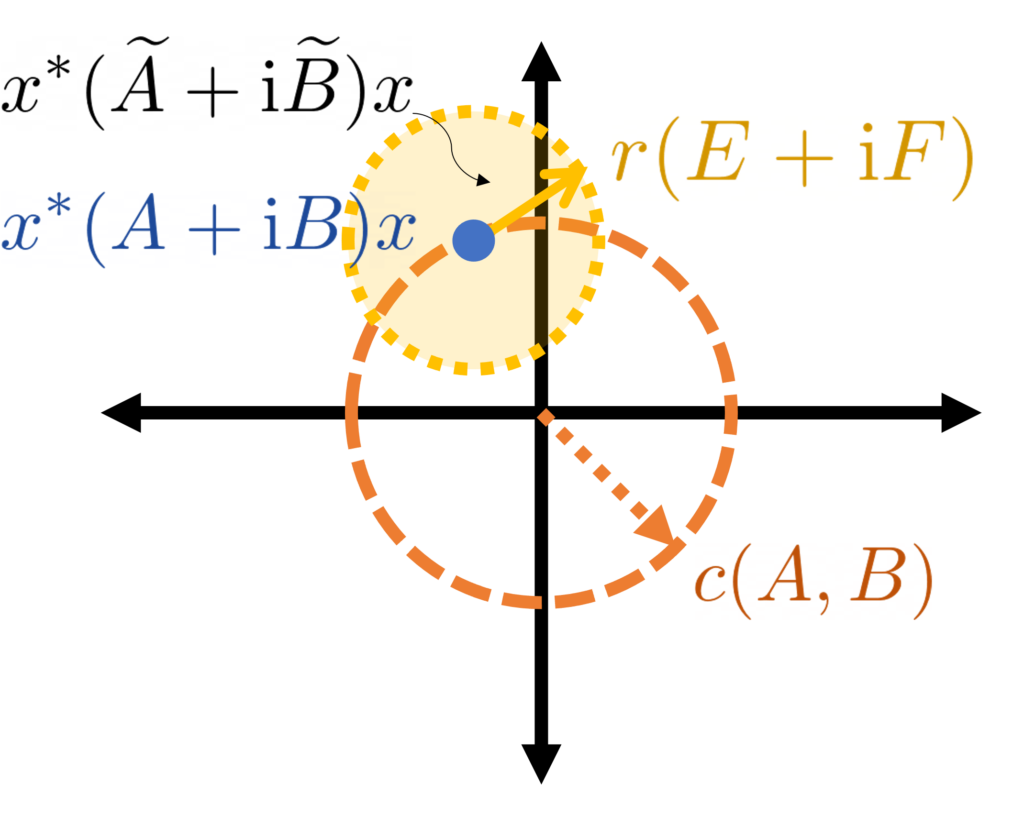
 might lie anywhere in a circle centered at
might lie anywhere in a circle centered at  for every
for every 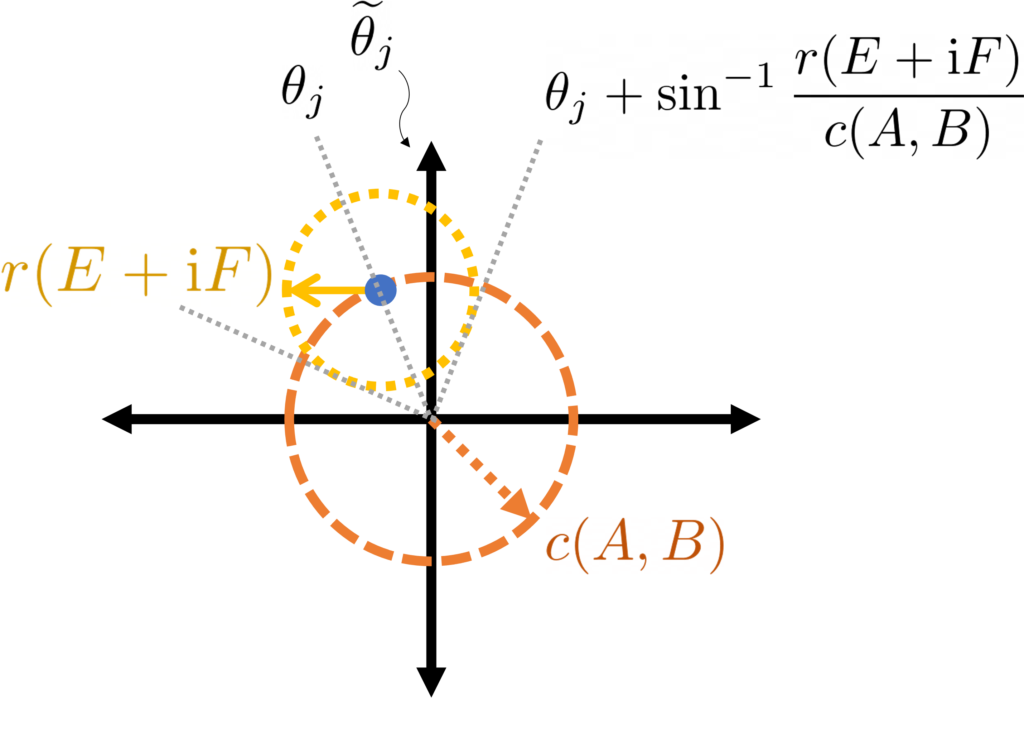
 .
. . An entirely analogous argument using the max-min variational principle (1) proves an identical lower bound, thus showing
. An entirely analogous argument using the max-min variational principle (1) proves an identical lower bound, thus showing

 , is much smaller than the definiteness of the original problem, measured by the Crawford number
, is much smaller than the definiteness of the original problem, measured by the Crawford number  and another hard-to-concisely describe quantity. In many cases, one has nothing better to do than to bound
and another hard-to-concisely describe quantity. In many cases, one has nothing better to do than to bound  , in which case the Crawford number has appeared again.
, in which case the Crawford number has appeared again. . The insight of the Mathias–Li theory is, in some sense, as simple as this: rather than using the fact that
. The insight of the Mathias–Li theory is, in some sense, as simple as this: rather than using the fact that  (as in Stewart’s analysis), use how far
(as in Stewart’s analysis), use how far  —the eigenvectors are thus made “
—the eigenvectors are thus made “ . In this way, just taking the eigenvector
. In this way, just taking the eigenvector  and
and  are diagonal matrices with entries
are diagonal matrices with entries  and
and  respectively. With this in mind, it can make our lives a lot easy to just do a change of variables
respectively. With this in mind, it can make our lives a lot easy to just do a change of variables  and
and  (which in turn sends
(which in turn sends  and
and  ). The change of variables
). The change of variables  . Further, note the following variational characterization of the spectral radius
. Further, note the following variational characterization of the spectral radius  . Plugging these two facts together yields
. Plugging these two facts together yields  .
. , which can be arbitrarily large.
, which can be arbitrarily large. and
and  the
the  and
and  where
where  .
.
 is the associated eigenangle, then this circle is subtended by arcs with angles
is the associated eigenangle, then this circle is subtended by arcs with angles
 were guaranteed to lie in these arcs (i.e.,
were guaranteed to lie in these arcs (i.e.,  ). Unfortunately this is not necessarily the case. If one
). Unfortunately this is not necessarily the case. If one  . In particular, this means that
. In particular, this means that  in decreasing order
in decreasing order  , we hace
, we hace  . An entirely analogous argument will give an upper bound, yielding
. An entirely analogous argument will give an upper bound, yielding
 . The trick will be to use the max-min characterization (1) with the subspace
. The trick will be to use the max-min characterization (1) with the subspace  . Churning through a couple inequalities in quick fashion,
. Churning through a couple inequalities in quick fashion,
 denotes the
denotes the  , it in particular holds for the set of indices which makes
, it in particular holds for the set of indices which makes  the largest. Thus,
the largest. Thus,  .
. by its minimum over all
by its minimum over all  (and
(and  )
)

 ; as Mathias and Li show
; as Mathias and Li show  .
. and
and  . (The precise instantiation of “sufficiently well-separated” requires some tedious algebra; if you’re interested, see
. (The precise instantiation of “sufficiently well-separated” requires some tedious algebra; if you’re interested, see 
 for a sufficiently small perturbation. In fact, a result of this form is nearly as good as one could hope for.
for a sufficiently small perturbation. In fact, a result of this form is nearly as good as one could hope for. , so we know for sufficiently small perturbations we have
, so we know for sufficiently small perturbations we have  and
and 
 numbers
numbers  rather than our full data set of 365,000 temperature values.
rather than our full data set of 365,000 temperature values.  with 1000 rows, one for each station, and 365 columns, one for each day of the year. The entry
with 1000 rows, one for each station, and 365 columns, one for each day of the year. The entry  corresponding to station
corresponding to station 
 for ease of discussion. When presented in this linear algebraic form, it’s less obvious in what way
for ease of discussion. When presented in this linear algebraic form, it’s less obvious in what way  . This example is suggestive that
. This example is suggestive that  is a weighted sum of the form
is a weighted sum of the form  , where
, where  are scalars
are scalars . If
. If  columns is guaranteed to be linearly dependent. Similarly, the row rank is defined to be the maximum size of any linearly independent collection of rows taken from
columns is guaranteed to be linearly dependent. Similarly, the row rank is defined to be the maximum size of any linearly independent collection of rows taken from  .
. entries. How can this be done?
entries. How can this be done? , where
, where  is an
is an  matrix and
matrix and  matrix. In other words,
matrix. In other words,  with
with  numbers down from
numbers down from  without ever forming
without ever forming  . Collect these
. Collect these  . But since the columns of
. But since the columns of  of
of  , where we suggestively use the labels
, where we suggestively use the labels  for the scalar multiples in our linear combination. Collecting these coefficients into a matrix
for the scalar multiples in our linear combination. Collecting these coefficients into a matrix  , we have constructed a factorization
, we have constructed a factorization  where
where  , and
, and  . With this definition, we can pick an arbitrary column basis
. With this definition, we can pick an arbitrary column basis  .
. where
where  and
and  and
and  is a (possibly rectangular) diagonal matrix with nonnegative, descending diagonal entries
is a (possibly rectangular) diagonal matrix with nonnegative, descending diagonal entries  . These diagonal entries are referred to as the singular values of the matrix
. These diagonal entries are referred to as the singular values of the matrix  (note that the remaining rows of
(note that the remaining rows of  for a vector
for a vector  . This reduces the operation count down from
. This reduces the operation count down from  operations to
operations to  operations using the rank factorization. As a general rule of thumb, when we have something expressed as a rank factorization, we can usually expect to reduce our operation count (and storage costs) from something proportional to
operations using the rank factorization. As a general rule of thumb, when we have something expressed as a rank factorization, we can usually expect to reduce our operation count (and storage costs) from something proportional to  .
. operations (expressed in
operations (expressed in  operations even to write down the matrices
operations even to write down the matrices  , where
, where  and
and  are
are  is a
is a  where
where  operations.
operations. and
and  . Reader beware: We call the “
. Reader beware: We call the “ and
and  , as we have already used the letter
, as we have already used the letter  .
. .
.  and
and  .
.  operations.
operations. operations.
operations. , a significant improvement over the
, a significant improvement over the  so
so  is
is  .
. at all costs. Instead, compute with the matrices
at all costs. Instead, compute with the matrices  where
where  by computing an
by computing an  , where
, where  factorization from scratch.
factorization from scratch.

 and
and  denote the
denote the  . (This involves solving
. (This involves solving  to compute each column
to compute each column  of
of  from each column
from each column  of
of  .) We then compute an LU factorization of the much smaller
.) We then compute an LU factorization of the much smaller  . Finally, we use our
. Finally, we use our  , from which our solution
, from which our solution  is given by
is given by
 to an operation count of
to an operation count of  which is dramatically better than the
which is dramatically better than the  operation count of recomputing the LU factorization from scratch.
operation count of recomputing the LU factorization from scratch. . An operation count like this still represents a dramatic improvement over the operation count
. An operation count like this still represents a dramatic improvement over the operation count  . The solution in this case is straightforward: approximate our high-rank matrix with a low-rank one, which we express in algorithmically useful form as a rank factorization.
. The solution in this case is straightforward: approximate our high-rank matrix with a low-rank one, which we express in algorithmically useful form as a rank factorization. which approximates
which approximates  and then throw away all but the first
and then throw away all but the first  . If
. If  is small. There might be many different ways of measuring the size of the error, but we have to insist on a couple of properties on our norm
is small. There might be many different ways of measuring the size of the error, but we have to insist on a couple of properties on our norm  , then the norm of
, then the norm of  should be
should be  . A list of the properties we require a norm to have are listed on the
. A list of the properties we require a norm to have are listed on the  and the
and the  , which measures the largest singular value.
, which measures the largest singular value. denote the
denote the 

 are the singular values of
are the singular values of  of position
of position  the time representing the
the time representing the  . As discussed in my article on
. As discussed in my article on 

 from a smooth function
from a smooth function  for points
for points  and
and  . Then we can expect that
. Then we can expect that  and
and  , then an
, then an  with
with  and
and  will be low-rank in the sense that it can be approximated to accuracy
will be low-rank in the sense that it can be approximated to accuracy  or the size of the domains
or the size of the domains ![[0,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4019f7e1f59924f7600fd76da233c29e_l3.png "Rendered by QuickLaTeX.com") is to pick 100 equispaced points
is to pick 100 equispaced points  on this interval and output the Riemann sum
on this interval and output the Riemann sum  . A randomized algorithm is simply an algorithm which uses, in some form, randomly chosen quantities. For instance, a randomized algorithm
. A randomized algorithm is simply an algorithm which uses, in some form, randomly chosen quantities. For instance, a randomized algorithm uniformly distributed in the interval
uniformly distributed in the interval  . To help to distinguish, an algorithm which does not use randomness is called a deterministic algorithm.
. To help to distinguish, an algorithm which does not use randomness is called a deterministic algorithm. operations
operations  operations
operations integers to sort in increasing order, quicksort works by selecting an element to be the pivot and then divides the elements of the list into groups larger and smaller than the pivot. One then recurses on the two groups, ending up with a sorted list.
integers to sort in increasing order, quicksort works by selecting an element to be the pivot and then divides the elements of the list into groups larger and smaller than the pivot. One then recurses on the two groups, ending up with a sorted list. time, which is asymptotically fast as this problem could possibly be solved since any median-finding algorithm must in general look at the entire list.
time, which is asymptotically fast as this problem could possibly be solved since any median-finding algorithm must in general look at the entire list. expected runtime
expected runtime is the fastest possible runtime for any
is the fastest possible runtime for any  algorithm. If one feeds in a list in a random order, the first-element pivot selection has a
algorithm. If one feeds in a list in a random order, the first-element pivot selection has a  , then we can still come up with an ordering of the input list that makes the algorithm run in time
, then we can still come up with an ordering of the input list that makes the algorithm run in time  .
. which solve the problem. (Less formally, we assign each possible algorithm a probability of picking it and pick one subject to these probabilities.) Once your opponent sees your randomized algorithm (equivalently, the distribution
which solve the problem. (Less formally, we assign each possible algorithm a probability of picking it and pick one subject to these probabilities.) Once your opponent sees your randomized algorithm (equivalently, the distribution  and present it to your algorithm.
and present it to your algorithm. with some distribution
with some distribution  to counter their strategy.
to counter their strategy.![\begin{equation*} \mathbb{E}_{I\sim Q} \left[ C(A_{\rm best},I) \right] \le \mathbb{E}_{A\sim P} \left[ \mathbb{E}_{I\sim Q} \left[ C(A, I) \right] \right]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-55ccc5c1f35b1e073bb8ccb0674706e3_l3.png "Rendered by QuickLaTeX.com")
 and
and  denote the
denote the ![\begin{equation*} \mathbb{E}_{A\sim P} \left[ \mathbb{E}_{I\sim Q} \left[ C(A, I) \right] \right] \le \mathbb{E}_{A\sim P} \left[ C(A,I_{\rm worst}) \right]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-08306ab131eefc536da92e60daacd796_l3.png "Rendered by QuickLaTeX.com")
![\begin{equation*} \mathbb{E}_{I\sim Q} \left[ C(A_{\rm best},I) \right] \le \mathbb{E}_{A\sim P} \left[ C(A,I_{\rm worst}) \right]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-048075ac1ea1ace95f8d86e6c79a981c_l3.png "Rendered by QuickLaTeX.com")
![\max_{\mbox{input distributions } Q} \min_{\mbox{algorithms }A} \mathbb{E}_{I\sim Q} [C(A,I)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6e5056cc57c21521a44f1bae4fa64f69_l3.png "Rendered by QuickLaTeX.com")
![=\min_{\mbox{randomized algorithms }P} \max_{\mbox{inputs }I}\mathbb{E}_{A\sim P} \left[ C(A,I) \right].](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9651554bb21521de67225ea9a22d15e6_l3.png "Rendered by QuickLaTeX.com") This nontrivial fact is a consequence of the full power of the
This nontrivial fact is a consequence of the full power of the  . If one in addition allows randomness, we call this
. If one in addition allows randomness, we call this  .
. .
. : all problems that can be tractably solved with randomness can tractably be solved without. There is
: all problems that can be tractably solved with randomness can tractably be solved without. There is 
 .
. minimal rank completion problem, which we will spend most of the remainder of this post discussing.
minimal rank completion problem, which we will spend most of the remainder of this post discussing.
 , to be the
, to be the  , one can extract a
, one can extract a  of
of 
 , as is exemplified when
, as is exemplified when  , we must also account for rows of
, we must also account for rows of  which cannot be written as linear combinations of the rows above, no matter how
which cannot be written as linear combinations of the rows above, no matter how  such rows, leading to the bound
such rows, leading to the bound
 the minimal rank for this completion problem.
the minimal rank for this completion problem. and
and  of the matrices
of the matrices  is again a vector subspace, and we choose a basis
is again a vector subspace, and we choose a basis  for it. The subspace
for it. The subspace  to it so that the enlarged matrix
to it so that the enlarged matrix  forms a basis for
forms a basis for  to add to
to add to  forms a basis for
forms a basis for  forms a basis for
forms a basis for  such that
such that  —in fact, this exact
—in fact, this exact  . Since
. Since  . Thus, doing the same with
. Thus, doing the same with 
 for the intersection of
for the intersection of  . We then extend these to bases
. We then extend these to bases  and
and  for
for  and
and  . From here, we derive the existence of matrix factorizations analogous to Eq. (4):
. From here, we derive the existence of matrix factorizations analogous to Eq. (4):
 . Applying this fact to the bases
. Applying this fact to the bases  and
and  for
for  such that
such that
 achieves the minimum possible rank
achieves the minimum possible rank  using the building blocks we’ve already built
using the building blocks we’ve already built
 entries whose value we have yet to determine. For us to have a hope of representing the matrix
entries whose value we have yet to determine. For us to have a hope of representing the matrix  into our rank factorization. Doing exactly this, we get the partially specified rank factorization
into our rank factorization. Doing exactly this, we get the partially specified rank factorization
 . From Eqs. (5) and (6), we deduce that
. From Eqs. (5) and (6), we deduce that  . Thus, we can fill in more entries:
. Thus, we can fill in more entries:
 and
and  to these free variables, we conclude that
to these free variables, we conclude that 

 given
given  is not universal. It is common to omit the factor in (1) and replace the
is not universal. It is common to omit the factor in (1) and replace the  in Eq. (2) with a
in Eq. (2) with a  . We prefer this convention as it makes the DFT a
. We prefer this convention as it makes the DFT a 
 is called the discrete Fourier transform (DFT) of
is called the discrete Fourier transform (DFT) of  . The FFT is just one possible algorithm to evaluate the DFT.
. The FFT is just one possible algorithm to evaluate the DFT.  for every integer
for every integer  for
for  . Then, in fact,
. Then, in fact,  , so indeed the right-hand side of Eq. (2) is indeed a “polynomial” in the “variables”
, so indeed the right-hand side of Eq. (2) is indeed a “polynomial” in the “variables”  and
and 

 of a trigonometric polynomial representation of
of a trigonometric polynomial representation of  represents the intensity of each pitch comprising the chord. An audio engineer could, for example, compute a Fourier series for a piece of music and zero out Fourier coefficients, thus reducing the amount of data needed to store a piece of music. This idea is indeed part of the way audio compression standards like
represents the intensity of each pitch comprising the chord. An audio engineer could, for example, compute a Fourier series for a piece of music and zero out Fourier coefficients, thus reducing the amount of data needed to store a piece of music. This idea is indeed part of the way audio compression standards like  , then we have that
, then we have that  . Recall the crucial fact from linear algebra that
. Recall the crucial fact from linear algebra that  for some
for some  . (We will omit the subscript
. (We will omit the subscript  . Then we have that
. Then we have that  . Comparing with Eq. (1), we see that
. Comparing with Eq. (1), we see that  . Let us define
. Let us define  . Thus, we can write the matrix
. Thus, we can write the matrix 
 in this discussion, but what will follow will generalize in a straightforward way to
in this discussion, but what will follow will generalize in a straightforward way to  ), we have
), we have

 is twenty-one eighths of a turn or simply just
is twenty-one eighths of a turn or simply just  turns. Thus
turns. Thus  and more generally
and more generally  . This allows us to simplify as follows:
. This allows us to simplify as follows:
 represents a quarter turn of the circle. This fact leads to the surprising observation we can actually find the DFT matrix
represents a quarter turn of the circle. This fact leads to the surprising observation we can actually find the DFT matrix  for
for  hidden inside the DFT matrix
hidden inside the DFT matrix  for
for  :
:
 sub-block is precisely
sub-block is precisely  (called the
(called the  , we have
, we have
 :
:
 represent the even-indexed entries of
represent the even-indexed entries of  the odd-indexed entries. Thus, we see that we can evaluate
the odd-indexed entries. Thus, we see that we can evaluate  by evaluating the two expressions
by evaluating the two expressions  and
and  . We have broken our problem into two smaller problems, which we then recombine into a solution of our original problem.
. We have broken our problem into two smaller problems, which we then recombine into a solution of our original problem. and
and  . Performing this process one more time, we need to evaluate expressions of the form
. Performing this process one more time, we need to evaluate expressions of the form  , which are simply given by
, which are simply given by  since the matrix
since the matrix  is just a
is just a  matrix whose single entry is
matrix whose single entry is  where
where  and
and  . Next, we combine these computations to evaluate
. Next, we combine these computations to evaluate 
 using the FFT can be determined by solving a certain
using the FFT can be determined by solving a certain  be the number of operations required by the FFT. Then the cost of computing
be the number of operations required by the FFT. Then the cost of computing  operations, in computer science language
operations, in computer science language refers to
refers to  operations is stating that, more or less, the algorithm takes less than some multiple of
operations is stating that, more or less, the algorithm takes less than some multiple of  operations to complete.
operations to complete. and
and for
for  , each of which requires
, each of which requires  operations.
operations.
 operations. This is a dramatic improvement of the
operations. This is a dramatic improvement of the  operations to compute
operations to compute  , where
, where  matrix,
matrix,  matrix,
matrix,  , and
, and  denotes the
denotes the  matrix defined as the
matrix defined as the 
 matrix and compute the matrix-vector product
matrix and compute the matrix-vector product  directly, but this takes a hefty
directly, but this takes a hefty  operations.
operations. . In the FFT, we needed to rearrange the columns of the DFT matrix
. In the FFT, we needed to rearrange the columns of the DFT matrix  and
and  of length
of length  and
and  respectively so that our matrix vector product can be written as
respectively so that our matrix vector product can be written as
 which takes time
which takes time  in total. Next, we compute each component
in total. Next, we compute each component  by using the formula
by using the formula
 operations to compute all the
operations to compute all the  ‘s. This leads to a total operation count of
‘s. This leads to a total operation count of  for computing the matrix-vector product
for computing the matrix-vector product  and
and  matrices
matrices  . The algorithm we presented is equivalent to evaluating this matrix triple product in the order
. The algorithm we presented is equivalent to evaluating this matrix triple product in the order  . This shows that this algorithm could be further accelerated using
. This shows that this algorithm could be further accelerated using 
 operations, just like the FFT.
operations, just like the FFT.
 . Let us by considering a special case of a Toeplitz matrix, a
. Let us by considering a special case of a Toeplitz matrix, a 
 is given by
is given by
 where
where  . This gives a fast algorithm to compute
. This gives a fast algorithm to compute  .
. of two functions
of two functions  on the real line. It is an important fact that
on the real line. It is an important fact that  (up to a possible normalizing constant).
(up to a possible normalizing constant).
 of the Toeplitz matrix Eq. (15) by
of the Toeplitz matrix Eq. (15) by  .
.  exactly equal to a power of two. This is useful because it allows us to compute matrix-vector products
exactly equal to a power of two. This is useful because it allows us to compute matrix-vector products  with the power-of-two FFT described above, which we know is fast.
with the power-of-two FFT described above, which we know is fast.
 and then discarding everything but the first entries of
and then discarding everything but the first entries of  to obtain
to obtain  th entry
th entry  . We now employ a clever trick. Let
. We now employ a clever trick. Let  . Then, defining
. Then, defining  , we have that
, we have that  , which means
, which means  , where the product
, where the product  , down from proportional to
, down from proportional to  th entry
th entry  .
. being replaced by
being replaced by  . The columns of the matrices
. The columns of the matrices  and
and  .
. and
and  . This follows from the calculations
. This follows from the calculations  and
and  . In other words, the nonzero singular values of
. In other words, the nonzero singular values of  or
or  . (These formulas are valid for invertible square matrices
. (These formulas are valid for invertible square matrices  of
of  and
and  may not be orthogonal matrices with, for example,
may not be orthogonal matrices with, for example,  not even close to the identity matrix. One can, of course, orthogonalize the computed
not even close to the identity matrix. One can, of course, orthogonalize the computed 
 are precisely plus-or-minus the singular values of
are precisely plus-or-minus the singular values of 
 and thus
and thus  for
for  account for all the nonzero eigenvalues of
account for all the nonzero eigenvalues of  is linear.
is linear. , an
, an  (respectively,
(respectively,  ), then
), then