In this post, we’ll talk about Markov chains, a useful and general model of a random system evolving in time.
PageRank
To see how Markov chains can be useful in practice, we begin our discussion with the famous PageRank problem. The goal is assign a numerical ranking to each website on the internet measuring how important it is. To do this, we form a mathematical model of an internet user randomly surfing the web. The importance of each website will be measured by the amount of times this user visits each page.
The PageRank model of an internet user is as follows: Start the user at an arbitrary initial website  . At each step, the user makes one of two choices:
. At each step, the user makes one of two choices:
- With 85% probability, the user follows a random link on their current website.
- With 15% probability, the user gets bored and jumps to a random website selected from the entire internet.
As with any mathematical model, this is a riduculously oversimplified description of how a person would surf the web. However, like any good mathematical model, it is useful. Because of the way the model is designed, the user will spend more time on websites with many incoming links. Thus, websites with many incoming links will be rated as important, which seems like a sensible choice.
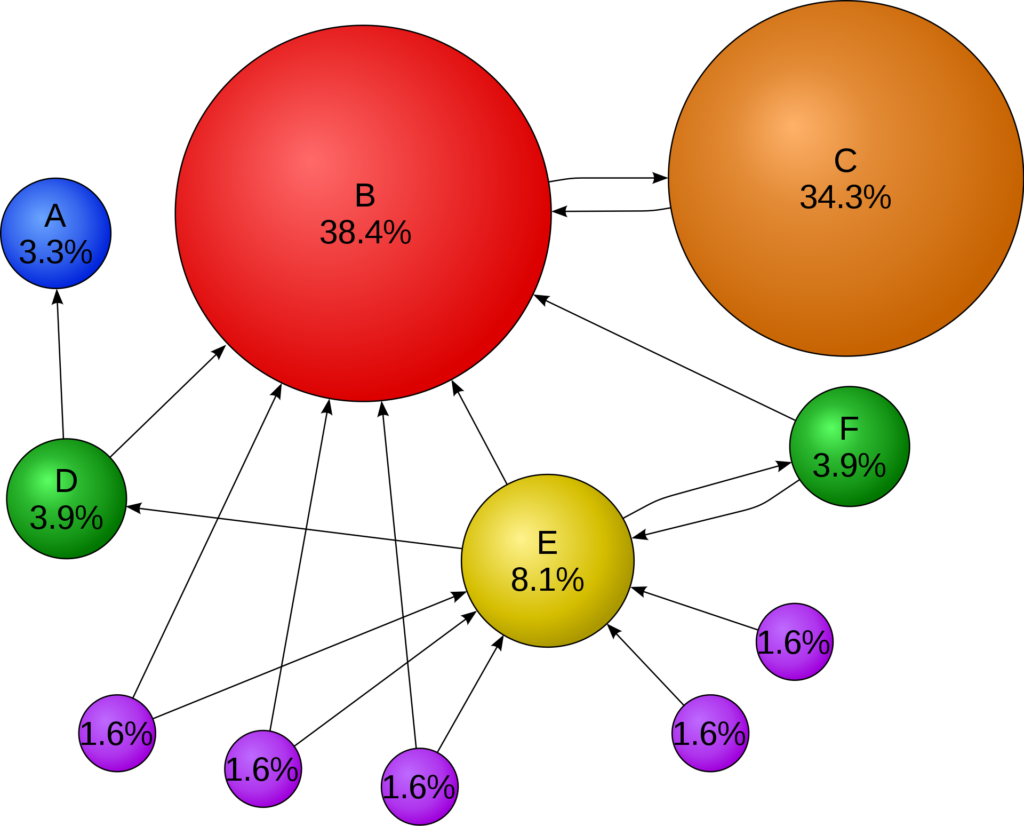
An example of the PageRank distribution for a small internet is shown below. As one would expect, the surfer spends a large part of their time on website B, which has many incoming links. Interestingly, the user spends almost as much of their time on website C, whose only links are to and from B. Under the PageRank model, a website is important if it is linked to by an important website, even if that is the only website linking to it.
Markov Chains in General
Having seen one Markov chain, the PageRank internet surfer, let’s talk about Markov chains in general. A (time-homogeneous) Markov chain consists of two things: a set of states and probabilities for transitioning between states:
- Set of states. For this discussion, we limit ourselves to Markov chains which can only exist in finitely many different states. To simplify our discussion, label the possible states using numbers
 .
. - Transition probabilities. The definining property of a (time-homogeneous) Markov chain is that, at any point in time
 , if the state is
, if the state is  , the probability of moving to state
, the probability of moving to state  is a fixed number
is a fixed number  . In particular, the probability of moving from to does not depend on the time or the past history of the chain before time ; only the value of the chain at time matters.
. In particular, the probability of moving from to does not depend on the time or the past history of the chain before time ; only the value of the chain at time matters.
Denote the state of the Markov chain at times  by
by  . Note that the states are random quantities. We can write the Markov chain property using the language of conditional probability:
. Note that the states are random quantities. We can write the Markov chain property using the language of conditional probability:
![\[\mathbb{P} \{ x_{n+1} = j \mid x_n = i,x_{n-1}=a_{n-1},\ldots,x_0=a_0\} = \mathbb{P}\{x_{n+1} = j \mid x_n = i\} = P_{ij}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d1fa5c29673402a40f530434ec99f998_l3.png "Rendered by QuickLaTeX.com")
This equation states that the probability that the system is in state at time  given the entire history of the system depends only on the value
given the entire history of the system depends only on the value  of the chain at time . This probability is the transition probability .
of the chain at time . This probability is the transition probability .
Let’s see how the PageRank internet surfer fits into this model:
- Set of states. Here, the set of states are the websites, which we label
 .
. - Transition probabilities. Consider two websites and . If does not have a link to , then the only way of going from to is if the surfer randomly gets bored (probability 15%) and picks website to visit at random (probability
 ). Thus,
). Thus, (
Suppose instead that )
) ![\[P_{ij} = \frac{0.15}{m}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e9cb5a0e824c976c19d2d73c1f45714b_l3.png "Rendered by QuickLaTeX.com") does link to and has
does link to and has  outgoing links. Then, in addition to the
outgoing links. Then, in addition to the  probability computed before, user has an 85% percent chance of following a link and a
probability computed before, user has an 85% percent chance of following a link and a  chance of picking as that link. Thus,
chance of picking as that link. Thus, (
 )
) ![\[P_{ij} = \frac{0.85}{d_i} + \frac{0.15}{m}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-51a6f3c00da58822f6465db8458a1f8c_l3.png "Rendered by QuickLaTeX.com")
Markov Chains and Linear Algebra
For a non-random process  , we can understand the processes evolution by determining its state
, we can understand the processes evolution by determining its state  at every point in time . Since Markov chains are random processes, it is not enough to track the state
at every point in time . Since Markov chains are random processes, it is not enough to track the state  of the process at every time . Rather, we must understand the probability distribution of the state at every point in time .
of the process at every time . Rather, we must understand the probability distribution of the state at every point in time .
It is customary in Markov chain theory to represent a probability distribution on the states  by a row vector
by a row vector  .1To really emphasize that probability distributions are row vectors, we shall write them as transposes of column vectors. So
.1To really emphasize that probability distributions are row vectors, we shall write them as transposes of column vectors. So  is a column vector but represents the probability distribution as is a row vector. The th entry
is a column vector but represents the probability distribution as is a row vector. The th entry  stores the probability that the system is in state . Naturally, as is a probability distribution, its entries must be nonnegative (
stores the probability that the system is in state . Naturally, as is a probability distribution, its entries must be nonnegative ( for every ) and add to one (
for every ) and add to one ( ).
).
Let  denote the probability distributions of the states
denote the probability distributions of the states  . It is natural to ask: How are the distributions related to each other? Let’s answer this question.
. It is natural to ask: How are the distributions related to each other? Let’s answer this question.
The probability that  is in state is the th entry of
is in state is the th entry of  :
:
![\[\rho^{(n+1)}_j = \mathbb{P} \{x_{n+1} = j\}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d8705a90b65f869a58cd432bbc884cfa_l3.png "Rendered by QuickLaTeX.com")
To compute this probability, we break into cases based on the value of the process at time : either  or
or  or … or
or … or  ; only one of these cases can be true at once. When we have an “or” of random events and these events are mutually exclusive (only can be true at once), then the probabilities add:
; only one of these cases can be true at once. When we have an “or” of random events and these events are mutually exclusive (only can be true at once), then the probabilities add:
![\[\rho^{(n+1)}_j = \mathbb{P} \{x_{n+1} = j\} = \sum_{i=1}^m \mathbb{P} \{x_{n+1} = j, x_n = i\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-448c23761c566dc3504d60b57f1ac8d1_l3.png "Rendered by QuickLaTeX.com")
Now we use some conditional probability. The probability that  and is the probability that times the probability that conditional on . That is,
and is the probability that times the probability that conditional on . That is,
![\[\rho^{(n+1)}_j = \sum_{i=1}^m \mathbb{P} \{x_{n+1} = j, x_n = i\} = \sum_{i=1}^m \mathbb{P} \{x_n = i\} \mathbb{P}\{x_{n+1} = j \mid x_n = i\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9b27f4d81b8b51c36f578f7e00017d96_l3.png "Rendered by QuickLaTeX.com")
Now, we can simplify using our definitions. The probability that is just  and the probability of moving from to is . Thus, we conclude
and the probability of moving from to is . Thus, we conclude
![\[\rho_j^{(n+1)} = \sum_{i=1}^m \rho^{(n)}_i P_{ij} .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1a858376daf68ec5f2d816ef52e66997_l3.png "Rendered by QuickLaTeX.com")
Phrased in the language of linear algebra, we’ve shown
![\[\left(\rho^{(n+1)}\right)^\top = \left(\rho^{(n)}\right)^\top P \quad \text{for any } n = 0,1,2,\ldots.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9ae710d7c26dc66ed497cc8faf34cab3_l3.png "Rendered by QuickLaTeX.com")
That is, if we view the transition probabilities as comprising an  matrix
matrix  , then the distribution at time is obtained by multiplying the distribution at time by transition matrix . In particular, if we iterate this result, we obtain that the distribution at time is given by
, then the distribution at time is obtained by multiplying the distribution at time by transition matrix . In particular, if we iterate this result, we obtain that the distribution at time is given by
![\[\left(\rho^{(n)}\right)^\top = \left(\rho^{(n-1)}\right)^\top P = \left[\left(\rho^{(n-2)}\right)^\top P\right]P = \left(\rho^{(n-2)}\right)^\top P^2 = \cdots = \left(\rho^{(0)}\right)^\top P^n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3fddd821b8cdb4b89262b0c72ea68e55_l3.png "Rendered by QuickLaTeX.com")
Thus, the distribution at time is the distribution at time  multiplied by the th power of the transition matrix .
multiplied by the th power of the transition matrix .
Convergence to Stationarity
Let’s go back to our web surfer again. At time , we started our surfer at a particular website, say  . As such, the probability distribution2To keep notation clean going forward, we will drop the transposes off of probability distributions, except when working with them linear algebraically.
. As such, the probability distribution2To keep notation clean going forward, we will drop the transposes off of probability distributions, except when working with them linear algebraically.  at time is concentrated just on website , with no other website having any probability at all. In the first few steps, most of the probability will remain in the vacinity of website , in the websites linked to by and the websites linked to by the websites linked to by and so on. However, as we run the chain long enough, the surfer will have time to widely across the web and the probability distribution will become less and less influenced by the chain’s starting location. This motivates the following definition:
at time is concentrated just on website , with no other website having any probability at all. In the first few steps, most of the probability will remain in the vacinity of website , in the websites linked to by and the websites linked to by the websites linked to by and so on. However, as we run the chain long enough, the surfer will have time to widely across the web and the probability distribution will become less and less influenced by the chain’s starting location. This motivates the following definition:
Definition. A Markov chain satisfies the mixing property if the probability distributions
converge to a single fixed probability distribution
regardless of how the chain is initialized (i.e., independent of the starting distribution
The distribution for a mixing Markov chain is known as a stationary distribution because it does not change under the action of :
(St) ![\[\pi^\top = \pi^\top P. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-daa1101ffcda26584d5f843c12765158_l3.png "Rendered by QuickLaTeX.com")
To see this, recall the recurrence
![\[\left(\rho^{(n+1)}\right)^\top = \left(\rho^{(n)}\right)^\top P,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bcbc3e101491a613c1dcb40680a4ef3b_l3.png "Rendered by QuickLaTeX.com")
take the limit as  , and observe that both
, and observe that both  and
and  converge to
converge to  .
.
One of the basic questions in the theory of Markov chains is finding conditions under which the mixing property (or suitable weaker versions of it) hold. To answer this question, we will need the following definition:
A Markov chain is primitive if, after running the chain for some number
![\[\text{There exists $n$ such that, for any $i,j = 1,2,\ldots,m$, } \quad\mathbb{P}\{x_n = j \mid x_0 = i \} = (P^n)_{ij} > 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9796d227fdde3830779ea16f255b9b70_l3.png "Rendered by QuickLaTeX.com")
The fundamental theorem of Markov chains is that primitive chains satisfy the mixing property.
Theorem (fundamental theorem of Markov chains). Every primitive Markov chain is mixing. In particular, there exists one and only probability distribution
converge to
Let’s see an example of the fundamental theorem with the PageRank surfer. After  step, there is at least a
step, there is at least a  chance of moving from any website to any other website . Thus, the chain is primitive. Consequently, there is a unique stationary distribution , and the surfer will converge to this stationary distribution regardless of which website they start at.
chance of moving from any website to any other website . Thus, the chain is primitive. Consequently, there is a unique stationary distribution , and the surfer will converge to this stationary distribution regardless of which website they start at.
Going Backwards in Time
Often, it is helpful to consider what would happen if we ran a Markov chain backwards in time. To see why this is an interesting idea, suppose you run website  and you’re interested in where your traffic is coming from. One way of achieving this would be to initialize the Markov chain at and run the chain backwards in time. Rather than asking, “given I’m at now, where would a user go next?”, you ask “given that I’m at now, where do I expect to have come from?”
and you’re interested in where your traffic is coming from. One way of achieving this would be to initialize the Markov chain at and run the chain backwards in time. Rather than asking, “given I’m at now, where would a user go next?”, you ask “given that I’m at now, where do I expect to have come from?”
Let’s formalize this notion a little bit. Consider a primitive Markov chain with stationary distribution . We assume that we initialize this Markov chain in the stationary distribution. That is, we pick  as our initial distribution for . The time-reversed Markov chain
as our initial distribution for . The time-reversed Markov chain  is defined as follows: The probability
is defined as follows: The probability  of moving from to in the time-reversed Markov chain is the probability that I was at state one step previously given that I’m at state now:
of moving from to in the time-reversed Markov chain is the probability that I was at state one step previously given that I’m at state now:
![\[\mathbb{P} \{y_1 = j \mid y_0 = i\} = P^{\rm rev}_{ij} = \mathbb{P} \{ x_0 = j \mid x_1 = i \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-caeb5343a623f571314596e612b7315d_l3.png "Rendered by QuickLaTeX.com")
To get a nice closed form expression for the reversed transition probabilities , we can invoke Bayes’ theorem:
(Rev) ![\[P^{\rm rev}_{ij} = \mathbb{P} \{ x_0 = j \mid x_1 = i \} = \frac{\mathbb{P} \{x_0 = j\} \mathbb{P} \{x_1 = i \mid x_0 = j\}}{\mathbb{P} \{x_1 = i\}} = \frac{ \pi_j P_{ji}}{\pi_i}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1ffaf13a6eb4a6d8079f1ee0f7501d5d_l3.png "Rendered by QuickLaTeX.com")
The time-reversed Markov chain can be a strange beast. For the reversed PageRank surfer, for instance, follows links “upstream” traveling from the linked site to the linking site. As such, our hypothetical website owner could get a good sense of where their traffic is coming from by initializing the reversed chain  at their website and following the chain one step back.
at their website and following the chain one step back.
Reversible Markov Chains
We now have two different Markov chains: the original and its time-reversal. Call a Markov chain reversible if these processes are the same. That is, if the transition probabilities are the same:
![\[P^{\rm rev}_{ij} = P_{ij} \quad \text{for every } i,j=1,2,\ldots,m.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4d394cf7f8d6b8a6318c27c3e447403d_l3.png "Rendered by QuickLaTeX.com")
Using our formula (Rev) for the reversed transition probability, the reversibility condition can be written more concisely as
![\[\pi_i P_{ij} = \pi_j P_{ji}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d6dade4e26238f165b12b7d2891d3a95_l3.png "Rendered by QuickLaTeX.com")
This condition is referred to as detailed balance.3There is an abstruse—but useful—way of reformulating the detailed balance condition. Think of a vector  as defining a function on the set
as defining a function on the set  ,
,  . Letting
. Letting  denote a random variable drawn from the stationary distribution
denote a random variable drawn from the stationary distribution  , we can define a non-standard inner product on
, we can define a non-standard inner product on  :
: ![\langle f, g\rangle_{\pi} \coloneqq \mathbb{E}[f(x) g(x)] = \sum_{i=1}^m \pi_i f(i)g(i)](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c871929113a513c08da0e9fa24a8043_l3.png "Rendered by QuickLaTeX.com") . Then the Markov chain is reversible if and only if detailed balance holds if and only if is a self-adjoint operator on when equipped with the non-standard inner product
. Then the Markov chain is reversible if and only if detailed balance holds if and only if is a self-adjoint operator on when equipped with the non-standard inner product  . This more abstract characterization has useful consequences. For instance, by the spectral theorem, the transition matrix of a reversible Markov chain has real eigenvalues and supports a basis of orthonormal eigenvectors (in the inner product). In words, it states that a Markov chain is reversible if, when initialized in the stationary distribution , the flow of probability mass from to (that is,
. This more abstract characterization has useful consequences. For instance, by the spectral theorem, the transition matrix of a reversible Markov chain has real eigenvalues and supports a basis of orthonormal eigenvectors (in the inner product). In words, it states that a Markov chain is reversible if, when initialized in the stationary distribution , the flow of probability mass from to (that is,  ) is equal to the flow of probability mass from to (that is,
) is equal to the flow of probability mass from to (that is,  ).
).
Many interesting Markov chains are reversible. One class of examples are Markov chain models of physical and chemical processes. Since physical laws like classical and quantum mechanics are reversible under time, so too should we expect Markov chain models built from theories to be reversible.
Not every interesting Markov chain is reversible, however. Indeed, except in special cases, the PageRank Markov chain is not reversible. If links to but. does not link to , then the flow of mass from to will be higher than the flow from to .
Before moving on, we note one useful fact about reversible Markov chains. Suppose a reversible, primitive Markov chain satisfies the detailed balance condition with a probability distribution  :
:
![\[\sigma_i P_{ij} = \sigma_j P_{ji}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f1ef7062af77b1ad2efc526c3db8a63b_l3.png "Rendered by QuickLaTeX.com")
Then  is the stationary distribution of this chain. To see why, we check the stationarity condition
is the stationary distribution of this chain. To see why, we check the stationarity condition  . Indeed, for every ,
. Indeed, for every ,
![\[(\sigma^\top P)_j = \sum_{i=1}^m \sigma_i P_{ij} = \sum_{i=1}^m \sigma_j P_{ji} = \sigma_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-81a7b02ba8de821bb56ecee978ec346d_l3.png "Rendered by QuickLaTeX.com")
The second equality is detailed balance and the third equality is just the condition that the sum of the transition probabilities from to each is one. Thus, and is a stationary distribution for . But a primitive chain has only one stationary distribution , so .
Markov Chains as Algorithms
Markov chains are an amazingly flexible tool. One use of Markov chains is more scientific: Given a system in the real world, we can model it by a Markov chain. By simulating the chain or by studying its mathematical properties, we can hope to learn about the system we’ve modeled.
Another use of Markov chains is algorithmic. Rather than thinking of the Markov chain as modeling some real-world process, we instead design the Markov chain to serve a computationally useful end. The PageRank surfer is one example. We wanted to rank the importance of websites, so we designed a Markov chain to achieve this task.
One task we can use Markov chains to solve are sampling problems. Suppose we have a complicated probability distribution , and we want a random sample from —that is, a random quantity  such that
such that  for every . One way to achieve this goal is to design a primitive Markov chain with stationary distribution . Then, we run the chain for a large number of steps and use as an approximate sample from .
for every . One way to achieve this goal is to design a primitive Markov chain with stationary distribution . Then, we run the chain for a large number of steps and use as an approximate sample from .
To design a Markov chain with stationary distribution , it is sufficient to generate transition probabilities such that and satisfy the detailed balance condition. Then, we are guaranteed that is a stationary distribution for the chain. (We also should check the primitiveness condition, but this is often straightforward.)
Here is an effective way of building a Markov chain to sample from a distribution . Suppose that the chain is in state at time , . To choose the next state, we begin by sampling from a proposal distribution  . The proposal distribution can be almost anything we like, as long as it satisfies three conditions:
. The proposal distribution can be almost anything we like, as long as it satisfies three conditions:
- Probability distribution. For every , the transition probabilitie
 add to one:
add to one:  .
. - Bidirectional. If
 , then
, then  .
. - Primitive. The transition probabilities form a primitive Markov chain.
In order to sample from the correct distribution, we can’t just accept every proposal. Rather, given the proposal , we accept with probability
![\[\min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fb8ef7791b1d955015f1300e0eddfb8c_l3.png "Rendered by QuickLaTeX.com")
If we accept the proposal, the next state of our chain is . Otherwise, we stay where we are  . This Markov chain is known as a Metropolis–Hastings sampler.
. This Markov chain is known as a Metropolis–Hastings sampler.
For clarity, we list the steps of the Metropolis–Hastings sampler explicitly:
- Initialize the chain in any state and set
 .
. - Draw a proposal
 with from the proposal distribution,
with from the proposal distribution,  .
. - Compute the acceptance probability
![\[p_{\rm acc} := \min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e34abfe410068f7516cd1c5e50100c3c_l3.png "Rendered by QuickLaTeX.com")
- With probability
 , set
, set  . Otherwise, set
. Otherwise, set  .
. - Set
 and go back to step 2.
and go back to step 2.
To check that is a stationary distribution of the Metropolis–Hastings distribution, all we need to do is check detailed balance. Note that the probability of transitioning from to  under the Metropolis–Hastings sampler is the proposal probability times the acceptance probability:
under the Metropolis–Hastings sampler is the proposal probability times the acceptance probability:
![\[P_{ij} = T_{ij} \cdot \min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8c98e72078f3de5c0de43a1683f5d406_l3.png "Rendered by QuickLaTeX.com")
Detailed balance is confirmed by a short computation4Note that the detailed balance condition for  is always satisfied for any Markov chain
is always satisfied for any Markov chain  .
.
![\[\pi_i P_{ij} = \pi_i T_{ij} \cdot \min \left\{ 1 , \frac{\pi_j T_{ji}}{\pi_i T_{ij}} \right\} = \min \left\{ \pi_i T_{ij} , \pi_j T_{ji} \right\} = \pi_j P_{ji}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9ec53c4f48f0cc7bb8cc1d0cd58750f1_l3.png "Rendered by QuickLaTeX.com")
Thus the Metropolis–Hastings sampler has as stationary distribution.
Determinatal Point Processes: Diverse Items from a Collection
The uses of Markov chains in science, engineering, math, computer science, and machine learning are vast. I wanted to wrap up with one application that I find particularly neat.
Suppose I run a bakery and I sell  different baked goods. I want to pick out
different baked goods. I want to pick out  special items for a display window to lure customers into my store. As a first approach, I might pick my top- selling items for the window. But I realize that there’s a problem. All of my top sellers are muffins, so all of the items in my display window are muffins. My display window is doing a good job luring in muffin-lovers, but a bad job of enticing lovers of other baked goods. In addition to rating the popularity of each item, I should also promote diversity in the items I select for my shop window.
special items for a display window to lure customers into my store. As a first approach, I might pick my top- selling items for the window. But I realize that there’s a problem. All of my top sellers are muffins, so all of the items in my display window are muffins. My display window is doing a good job luring in muffin-lovers, but a bad job of enticing lovers of other baked goods. In addition to rating the popularity of each item, I should also promote diversity in the items I select for my shop window.
Here’s a creative solution to my display case problems using linear algebra. Suppose that, rather than just looking at a list of the sales of each item, I define a matrix  for my baked goods. In the
for my baked goods. In the  th entry
th entry  of my matrix, I write the number of sales for baked good . I populate the off-diagonal entries
of my matrix, I write the number of sales for baked good . I populate the off-diagonal entries  of my matrix with a measure of similarity between items and .5There are many ways of defining such a similarity matrix. Here is one way. Let
of my matrix with a measure of similarity between items and .5There are many ways of defining such a similarity matrix. Here is one way. Let  be the number ordered for each bakery item by a random customer. Set
be the number ordered for each bakery item by a random customer. Set  to be the correlation matrix of the random variables , with
to be the correlation matrix of the random variables , with  being the correlation between the random variables
being the correlation between the random variables  and
and  . The matrix has all ones on its diagonal. The off-diagonal entries measure the amount that items and tend to be purchased together. Let
. The matrix has all ones on its diagonal. The off-diagonal entries measure the amount that items and tend to be purchased together. Let  be a diagonal matrix where
be a diagonal matrix where  is the total sales of item . Set
is the total sales of item . Set  . By scaling by the diagonal matrix , the diagonal entries of represent the popularity of each item, whereass the off-diagonal entries still represent correlations, now scaled by popularity. So if and are both muffins, will be large. But if is a muffin and is a cookie, then will be small. For mathematical reasons, we require to be symmetric and positive definite.
. By scaling by the diagonal matrix , the diagonal entries of represent the popularity of each item, whereass the off-diagonal entries still represent correlations, now scaled by popularity. So if and are both muffins, will be large. But if is a muffin and is a cookie, then will be small. For mathematical reasons, we require to be symmetric and positive definite.
To populate my display case, I choose a random subset of items from my full menu of size according to the following strange probability distribution: The probability  of picking items
of picking items  is proportional to the determinant of the submatrix
is proportional to the determinant of the submatrix  . More specifically,
. More specifically,
(-DPP) ![\[\pi_S = \frac{\det A(S,S)}{\sum_{\text{all subsets $T$ of size $k$}} \det A(T,T)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-26714c8980fc33f439de8cf1e5a692eb_l3.png "Rendered by QuickLaTeX.com")
Here, we let denote the  submatrix of consisting of the entries appearing in rows and columns
submatrix of consisting of the entries appearing in rows and columns  . Such a random subset is known as a -determinantal point process (-DPP). (See this survey for more about DPPs.)
. Such a random subset is known as a -determinantal point process (-DPP). (See this survey for more about DPPs.)
To see why this makes any sense, let’s consider a simple example of  items and a display case of size
items and a display case of size  . Suppose I have three items: a pumpkin muffin, a chocolate chip muffin, and an oatmeal raisin cookies. Say the matrix looks like
. Suppose I have three items: a pumpkin muffin, a chocolate chip muffin, and an oatmeal raisin cookies. Say the matrix looks like
![\[A = \begin{bmatrix} 10 & 9 & 0 \\ 9 & 10 & 0 \\ 0 & 0 & 5 \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8bb20f6bff68d835397eabaf8a1cdc0_l3.png "Rendered by QuickLaTeX.com")
We see that both muffins are equally popular  and much more popular than the cookie
and much more popular than the cookie  . However, the two muffins are similar to each other and thus the corresponding submatrix has small determinant
. However, the two muffins are similar to each other and thus the corresponding submatrix has small determinant
![\[\det A(\{1,2\},\{1,2\}) = \det \twobytwo{10}{9}{9}{10} = 19.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7f74f9426bb0c808b9e4384a0a2a6c2d_l3.png "Rendered by QuickLaTeX.com")
By contrast, if the cookie is disimilar to each muffin and the determinant is higher
![\[\det A(\{1,3\},\{1,3\}) = \det A(\{2,3\},\{2,3\}) = \det \twobytwo{10}{0}{0}{5} = 50.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-812d8318cba1d4a937e92858658ff7e8_l3.png "Rendered by QuickLaTeX.com")
Thus, even though the muffins are more popular overall, choosing our display case from a  -DPP, we have a
-DPP, we have a  chance of choosing a muffin and a cookie for our display case. It is for this reason that we can say that a -DPP preferentially selects for diverse items.
chance of choosing a muffin and a cookie for our display case. It is for this reason that we can say that a -DPP preferentially selects for diverse items.
Is sampling from a -DPP the best way of picking items for my display case? How does it compare to other possible methods?6Another method I’m partial to for this task is randomly pivoted Cholesky sampling, which is computationally cheaper than -DPP sampling even with the Markov chain sampling approach to -DPP sampling that we will discuss shortly. These are interesting questions for another time. For now, let us focus our attention on a different question: How would you sample from a -DPP?
Determinantal Point Process by Markov Chains
Sampling from a -DPP is a hard computational problem. Indeed, there are  possible -element subspaces of a set of items. The number of possibilities gets large fast. If I have
possible -element subspaces of a set of items. The number of possibilities gets large fast. If I have  items and want to pick
items and want to pick  of them, there are already over 10 trillion possible combinations.
of them, there are already over 10 trillion possible combinations.
Markov chains offer one compelling way of sampling a -DPP. First, we need a proposal distribution. Let’s choose the simplest one we can think of:
Proposal for
. To generate a proposal, choose a uniformly random element
out of
and a uniformly random element
out of
without
obtained from
).
Now, we need to compute the Metropolis–Hastings acceptance probability
![\[p_{\rm acc} = \min \left\{ 1 , \frac{\pi_{S'} T_{S'S}}{\pi_{S} T_{SS'}} \right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5ff5280a20cfddd08b7917152edbfb68_l3.png "Rendered by QuickLaTeX.com")
For and which differ only by the addition of one element and the removal of another, the proposal probabilities  and
and  are both equal to
are both equal to  ,
,  . Using the formula for the probability of drawing from a -DPP, we compute that
. Using the formula for the probability of drawing from a -DPP, we compute that
![\[\frac{\pi_{S'}}{\pi_S} = \frac{\det A(S',S')}{\det A(S,S)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-26f4fdbc4d7624e49904e22703e8aaa7_l3.png "Rendered by QuickLaTeX.com")
Thus, the Metropolis–Hastings acceptance probability is just a ratio of determinants:
(Acc) ![\[p_{\rm acc} = \min \left\{ 1 , \frac{\pi_{S'} T_{S'S}}{\pi_{S} T_{SS'}} \right\} = \min \left\{ 1, \frac{\det A(S',S')}{\det A(S,S)} \right\}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-16c835576499e7fb3e6cf2c219732873_l3.png "Rendered by QuickLaTeX.com")
And we’re done. Let’s summarize our sampling algorithm:
- Choose an initial set
 arbitrarily and set .
arbitrarily and set . - Draw uniformly at random from
 .
. - Draw uniformly at random from
 .
. - Set
 .
. - With probability defined in (Acc), accept and set
 . Otherwise, set
. Otherwise, set  .
. - Set and go to step 2.
This is a remarkably simple algorithm to sample from a complicated distribution. And its fairly efficient as well. Analysis by Anari, Oveis Gharan, and Rezaei shows that, when you pick a good enough initial set , this sampling algorithm produces approximate samples from a -DPP in roughly  steps.7They actually use a slight variant of this algorithm where the acceptance probabilities (Acc) are reduced by a factor of two. Observe that this still has the correct stationary distribution because detailed balance continues to hold. The extra factor is introduced to ensure that the Markov chain is primitive. Remarkably, if is much smaller than , this Markov chain-based algorithm samples from a -DPP without even looking at all
steps.7They actually use a slight variant of this algorithm where the acceptance probabilities (Acc) are reduced by a factor of two. Observe that this still has the correct stationary distribution because detailed balance continues to hold. The extra factor is introduced to ensure that the Markov chain is primitive. Remarkably, if is much smaller than , this Markov chain-based algorithm samples from a -DPP without even looking at all  entries of the matrix !
entries of the matrix !
Upshot. Markov chains are a simple and general model for a state evolving randomly in time. Under mild conditions, Markov chains converge to a stationary distribution: In the limit of a large number of steps, the state of the system become randomly distributed in a way independent of how it was initialized. We can use Markov chains as algorithms to approximately sample from challenging distributions.
 triplets and checking whether they form a triangle is far too computationally costly for the billions of Facebook accounts. Somehow, we want to do much faster than this, and to achieve this speed we would be willing to settle for an estimate of the triangle count up to some error.
triplets and checking whether they form a triangle is far too computationally costly for the billions of Facebook accounts. Somehow, we want to do much faster than this, and to achieve this speed we would be willing to settle for an estimate of the triangle count up to some error. matrix where the
matrix where the  th entry of
th entry of  of the matrix
of the matrix  where
where  , and
, and  , which is twice the number of triangles incident on
, which is twice the number of triangles incident on  and
and  are both counted as paths of length 3 for a triangle consisting of
are both counted as paths of length 3 for a triangle consisting of  is counted thrice in the
is counted thrice in the  th, and
th, and  th entries of
th entries of 
 . We assume that we only have access to the matrix
. We assume that we only have access to the matrix  for a vector
for a vector  than the maximum possible amount of
than the maximum possible amount of  . Therefore, the matrix
. Therefore, the matrix  , we simply compute matrix–vector products with
, we simply compute matrix–vector products with  .
. -values, where each value
-values, where each value  and
and  occurs with equal
occurs with equal  probability. Then if one forms the expression
probability. Then if one forms the expression  . Since the entries of
. Since the entries of  and
and  are independent, the
are independent, the  is
is  and
and  is
is![\begin{equation*} \mathbb{E} \, x^\top M x = \sum_{i,j=1}^n M_{ij} \mathbb{E} [x_ix_j] = \sum_{i = 1}^n M_{ii} = \operatorname{tr}(M). \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-17a6a7cacc94a45f13853dc8f34f1e3a_l3.png "Rendered by QuickLaTeX.com")
 . Thus, the efficiently computable quantity
. Thus, the efficiently computable quantity  equals
equals  and compute the averaged trace estimator
and compute the averaged trace estimator
 remains an unbiased estimator for
remains an unbiased estimator for  , but with reduced variability. Quantitatively, the
, but with reduced variability. Quantitatively, the 
 .” In other words, concentration inequalities can provide quantitative estimates of the likely size of the error when a randomized algorithm is executed.
.” In other words, concentration inequalities can provide quantitative estimates of the likely size of the error when a randomized algorithm is executed. be any nonnegative random variable. Markov’s inequality states that the probability that
be any nonnegative random variable. Markov’s inequality states that the probability that  is bounded by the expected value of
is bounded by the expected value of  . In equations, we have
. In equations, we have
 . This small example shows both the power and the limitation of Markov’s inequality. On the negative side, our analysis suggests that we might have to wait as much as 100 times the average runtime for the algorithm to complete running with 99% probability; this large huge multiple of 100 seems quite pessimistic. On the other hand, we needed no information whatsoever about how the algorithm works to do this analysis. In general, Markov’s inequality cannot be improved without more assumptions on the random variable
. This small example shows both the power and the limitation of Markov’s inequality. On the negative side, our analysis suggests that we might have to wait as much as 100 times the average runtime for the algorithm to complete running with 99% probability; this large huge multiple of 100 seems quite pessimistic. On the other hand, we needed no information whatsoever about how the algorithm works to do this analysis. In general, Markov’s inequality cannot be improved without more assumptions on the random variable  :
:
 are
are  and variance
and variance  and let
and let  denote the average
denote the average

 . Therefore, by Chebyshev’s inequality,
. Therefore, by Chebyshev’s inequality,
 and are willing to tolerate a failure probability of
and are willing to tolerate a failure probability of  . Then setting the right-hand side of (5) to
. Then setting the right-hand side of (5) to 
 . Normal random variables have an exponentially small probability of being more than a few standard deviations above their mean, so it is natural to expect this should be true of
. Normal random variables have an exponentially small probability of being more than a few standard deviations above their mean, so it is natural to expect this should be true of 
 . Hoeffding’s inequality makes the assumption that the summands are bounded, say within an interval
. Hoeffding’s inequality makes the assumption that the summands are bounded, say within an interval ![[a,b]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-336bb22d4b2092329486008f6665b888_l3.png "Rendered by QuickLaTeX.com") .
.
 replaced by the potentially much larger quantity
replaced by the potentially much larger quantity .
. for every
for every  . We continue to denote
. We continue to denote 

 . We conclude that Bernstein’s inequality provides sharper bounds then Hoeffding’s inequality for smaller values of
. We conclude that Bernstein’s inequality provides sharper bounds then Hoeffding’s inequality for smaller values of  , except with failure probability at most
, except with failure probability at most 

![[\mu-B,\mu+B]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bada0c6b976891e43bcc4c3b7508e96e_l3.png "Rendered by QuickLaTeX.com") for every
for every  , then
, then
 samples are needed rather than proportional to
samples are needed rather than proportional to  . The fact that we need proportional to
. The fact that we need proportional to  samples to achieve error
samples to achieve error  to
to  , which is a huge improvement.
, which is a huge improvement. ; for small values of
; for small values of 
 is the
is the  is the
is the 
 . By the
. By the  and
and  where
where  and
and  are the
are the  is the
is the  so
so  and
and  . Since the
. Since the 
 denotes the
denotes the 

 and
and  . Since we often don’t know good bounds for
. Since we often don’t know good bounds for  , one should really use the trace estimator together with an
, one should really use the trace estimator together with an  down from
down from  .
. is an
is an 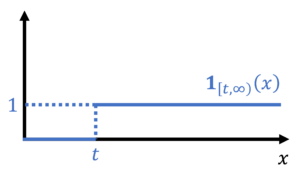
![\begin{equation*} \mathbb{P}\{X \ge t \} = \mathbb{E}[\mathbf{1}_{[t,\infty)}(X)]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-070609e7e86c3846db2eec810355d5f4_l3.png "Rendered by QuickLaTeX.com")
 by bounding its corresponding indicator function. In particular, we have the inequality
by bounding its corresponding indicator function. In particular, we have the inequality 
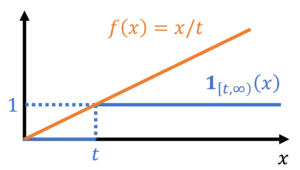
![\begin{equation*} \mathbb{P}\{ X \ge t \} = \mathbb{E}[\mathbf{1}_{[t,\infty)}(X)] \le \mathbb{E} \left[ \frac{X}{t} \right] = \frac{\mathbb{E} X}{t}. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49a753eda4e591b364f5d4f3e24a0b9b_l3.png "Rendered by QuickLaTeX.com")

 by (11). To obtain sharp and useful bounds on
by (11). To obtain sharp and useful bounds on  we seek bounding functions
we seek bounding functions  in (13) with three properties:
in (13) with three properties: ,
,  should be close to zero,
should be close to zero, ,
,  to be easily computable or boundable.
to be easily computable or boundable. or exponentials
or exponentials  for which we have hopes of computing or bounding
for which we have hopes of computing or bounding  (by increasing
(by increasing  ) to meet point 1 makes the function larger on
) to meet point 1 makes the function larger on  , detracting from our ability to achieve point 2. We shall eventually come up with a best-possible resolution to this dilemma by formulating this as an optimization problem to determine the best choice of the parameter
, detracting from our ability to achieve point 2. We shall eventually come up with a best-possible resolution to this dilemma by formulating this as an optimization problem to determine the best choice of the parameter  to obtain the best possible candidate function of the given form.
to obtain the best possible candidate function of the given form. satisfies the bound (13), and thus by (12),
satisfies the bound (13), and thus by (12),
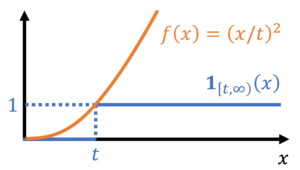
 , is a nonnegative random variable. Thus applying (14) gives
, is a nonnegative random variable. Thus applying (14) gives
 if, and only if,
if, and only if,  . Therefore, by Markov’s inequality,
. Therefore, by Markov’s inequality,
 , where
, where 

 ,
,  , and the
, and the 
 up to the sign of the parameter
up to the sign of the parameter  . Taking logarithms proves the additivity.
. Taking logarithms proves the additivity.
 and consider the cumulant generating function of
and consider the cumulant generating function of  . Then one can show the cumulant generating function bound
. Then one can show the cumulant generating function bound . We have
. We have  . Since the function
. Since the function  is
is  . Taking expectations, we have
. Taking expectations, we have  . One can show by comparing Taylor series that
. One can show by comparing Taylor series that  . Therefore, we have
. Therefore, we have  .
.



 :
:
 , we can apply a small trick. If we apply (20) to the summands
, we can apply a small trick. If we apply (20) to the summands  instead of
instead of 
 . The means of doing often this goes by the fancy name
. The means of doing often this goes by the fancy name 


 with mean zero and satisfying the bound
with mean zero and satisfying the bound  .
.
 denotes the average temperature of the station and
denotes the average temperature of the station and  denotes the maximum deviation above or below this station, signed so that it is warmer than average in the Northern hemisphere during June-August and colder-than-average in the Southern hemisphere during these months. The
denotes the maximum deviation above or below this station, signed so that it is warmer than average in the Northern hemisphere during June-August and colder-than-average in the Southern hemisphere during these months. The  is chosen so the hottest (or coldest) day in the year occurs at the
is chosen so the hottest (or coldest) day in the year occurs at the  numbers
numbers  rather than our full data set of 365,000 temperature values.
rather than our full data set of 365,000 temperature values.  with 1000 rows, one for each station, and 365 columns, one for each day of the year. The entry
with 1000 rows, one for each station, and 365 columns, one for each day of the year. The entry  corresponding to station
corresponding to station 
 for ease of discussion. When presented in this linear algebraic form, it’s less obvious in what way
for ease of discussion. When presented in this linear algebraic form, it’s less obvious in what way  . This example is suggestive that
. This example is suggestive that  is a weighted sum of the form
is a weighted sum of the form  , where
, where  are scalars
are scalars . If
. If  is the size of the largest possible subset of
is the size of the largest possible subset of  , then there is some sub-collection of
, then there is some sub-collection of  columns is guaranteed to be linearly dependent. Similarly, the row rank is defined to be the maximum size of any linearly independent collection of rows taken from
columns is guaranteed to be linearly dependent. Similarly, the row rank is defined to be the maximum size of any linearly independent collection of rows taken from  .
. matrix
matrix  entries. How can this be done?
entries. How can this be done? , where
, where  is an
is an  matrix and
matrix and  is an
is an  matrix. In other words,
matrix. In other words,  with
with  numbers down from
numbers down from  without ever forming
without ever forming  . Collect these
. Collect these  . But since the columns of
. But since the columns of  of
of  , where we suggestively use the labels
, where we suggestively use the labels  for the scalar multiples in our linear combination. Collecting these coefficients into a matrix
for the scalar multiples in our linear combination. Collecting these coefficients into a matrix  , we have constructed a factorization
, we have constructed a factorization  where
where  , and
, and  . With this definition, we can pick an arbitrary column basis
. With this definition, we can pick an arbitrary column basis  .
. where
where  and
and  are an
are an  . These diagonal entries are referred to as the singular values of the matrix
. These diagonal entries are referred to as the singular values of the matrix  (note that the remaining rows of
(note that the remaining rows of  for a vector
for a vector  . This reduces the operation count down from
. This reduces the operation count down from  operations to
operations to  operations using the rank factorization. As a general rule of thumb, when we have something expressed as a rank factorization, we can usually expect to reduce our operation count (and storage costs) from something proportional to
operations using the rank factorization. As a general rule of thumb, when we have something expressed as a rank factorization, we can usually expect to reduce our operation count (and storage costs) from something proportional to  .
. operations (expressed in
operations (expressed in  operations even to write down the matrices
operations even to write down the matrices  , where
, where  and
and  are
are  is a
is a  is a factorization
is a factorization  where
where  is an
is an  operations.
operations. and
and  . Reader beware: We call the “
. Reader beware: We call the “ and
and  , as we have already used the letter
, as we have already used the letter  .
. .
.  and
and  .
.  operations.
operations. operations.
operations. , a significant improvement over the
, a significant improvement over the  so
so  is
is  .
. at all costs. Instead, compute with the matrices
at all costs. Instead, compute with the matrices  where
where  by computing an
by computing an  , where
, where  factorization from scratch.
factorization from scratch.

 and
and  denote the
denote the  . (This involves solving
. (This involves solving  to compute each column
to compute each column  of
of  of
of  . Finally, we use our
. Finally, we use our  , from which our solution
, from which our solution  is given by
is given by
 to an operation count of
to an operation count of  which is dramatically better than the
which is dramatically better than the  operation count of recomputing the LU factorization from scratch.
operation count of recomputing the LU factorization from scratch. . An operation count like this still represents a dramatic improvement over the operation count
. An operation count like this still represents a dramatic improvement over the operation count  . The solution in this case is straightforward: approximate our high-rank matrix with a low-rank one, which we express in algorithmically useful form as a rank factorization.
. The solution in this case is straightforward: approximate our high-rank matrix with a low-rank one, which we express in algorithmically useful form as a rank factorization. which approximates
which approximates  and then throw away all but the first
and then throw away all but the first  which we will call
which we will call  . If
. If  is small. There might be many different ways of measuring the size of the error, but we have to insist on a couple of properties on our norm
is small. There might be many different ways of measuring the size of the error, but we have to insist on a couple of properties on our norm  , then the norm of
, then the norm of  should be
should be  . A list of the properties we require a norm to have are listed on the
. A list of the properties we require a norm to have are listed on the  and the
and the  , which measures the largest singular value.
, which measures the largest singular value. denote the
denote the 

 are the singular values of
are the singular values of  of position
of position  the time representing the
the time representing the  . As discussed in my article on
. As discussed in my article on 

 from a smooth function
from a smooth function  for points
for points  and
and  . Then we can expect that
. Then we can expect that  in the product of compact regions
in the product of compact regions  and
and  , then an
, then an  with
with  and
and  will be low-rank in the sense that it can be approximated to accuracy
will be low-rank in the sense that it can be approximated to accuracy  or the size of the domains
or the size of the domains  given
given  defined by the formula
defined by the formula is not universal. It is common to omit the factor in (1) and replace the
is not universal. It is common to omit the factor in (1) and replace the  in Eq. (2) with a
in Eq. (2) with a  . We prefer this convention as it makes the DFT a
. We prefer this convention as it makes the DFT a 
 is called the discrete Fourier transform (DFT) of
is called the discrete Fourier transform (DFT) of  . The FFT is just one possible algorithm to evaluate the DFT.
. The FFT is just one possible algorithm to evaluate the DFT.  is a periodic function defined on the integers with period
is a periodic function defined on the integers with period  for every integer
for every integer  for
for  . Then, in fact,
. Then, in fact,  , so indeed the right-hand side of Eq. (2) is indeed a “polynomial” in the “variables”
, so indeed the right-hand side of Eq. (2) is indeed a “polynomial” in the “variables”  and
and 

 of a periodic function
of a periodic function  of a trigonometric polynomial representation of
of a trigonometric polynomial representation of  represents the intensity of each pitch comprising the chord. An audio engineer could, for example, compute a Fourier series for a piece of music and zero out Fourier coefficients, thus reducing the amount of data needed to store a piece of music. This idea is indeed part of the way audio compression standards like
represents the intensity of each pitch comprising the chord. An audio engineer could, for example, compute a Fourier series for a piece of music and zero out Fourier coefficients, thus reducing the amount of data needed to store a piece of music. This idea is indeed part of the way audio compression standards like  , then we have that
, then we have that  . Recall the crucial fact from linear algebra that
. Recall the crucial fact from linear algebra that  for some
for some  . (We will omit the subscript
. (We will omit the subscript  by
by  . Then we have that
. Then we have that  . Comparing with Eq. (1), we see that
. Comparing with Eq. (1), we see that  . Let us define
. Let us define  . Thus, we can write the matrix
. Thus, we can write the matrix 
 in this discussion, but what will follow will generalize in a straightforward way to
in this discussion, but what will follow will generalize in a straightforward way to  ), we have
), we have

 is twenty-one eighths of a turn or simply just
is twenty-one eighths of a turn or simply just  turns. Thus
turns. Thus  and more generally
and more generally  . This allows us to simplify as follows:
. This allows us to simplify as follows:
 represents a quarter turn of the circle. This fact leads to the surprising observation we can actually find the DFT matrix
represents a quarter turn of the circle. This fact leads to the surprising observation we can actually find the DFT matrix  for
for  hidden inside the DFT matrix
hidden inside the DFT matrix  for
for  :
:
 sub-block is precisely
sub-block is precisely  (called the
(called the  , we have
, we have
 :
:
 represent the even-indexed entries of
represent the even-indexed entries of  the odd-indexed entries. Thus, we see that we can evaluate
the odd-indexed entries. Thus, we see that we can evaluate  by evaluating the two expressions
by evaluating the two expressions  and
and  . We have broken our problem into two smaller problems, which we then recombine into a solution of our original problem.
. We have broken our problem into two smaller problems, which we then recombine into a solution of our original problem. and
and  . Performing this process one more time, we need to evaluate expressions of the form
. Performing this process one more time, we need to evaluate expressions of the form  , which are simply given by
, which are simply given by  since the matrix
since the matrix  is just a
is just a  matrix whose single entry is
matrix whose single entry is  where
where  and
and  . Next, we combine these computations to evaluate
. Next, we combine these computations to evaluate 
 using the FFT can be determined by solving a certain
using the FFT can be determined by solving a certain  be the number of operations required by the FFT. Then the cost of computing
be the number of operations required by the FFT. Then the cost of computing  operations, in computer science language
operations, in computer science language refers to
refers to  operations is stating that, more or less, the algorithm takes less than some multiple of
operations is stating that, more or less, the algorithm takes less than some multiple of  operations to complete.
operations to complete. and
and for
for  , each of which requires
, each of which requires  operations.
operations.
 operations. This is a dramatic improvement of the
operations. This is a dramatic improvement of the  operations to compute
operations to compute  , where
, where  matrix,
matrix,  matrix,
matrix,  , and
, and  denotes the
denotes the  matrix defined as the
matrix defined as the 
 matrix and compute the matrix-vector product
matrix and compute the matrix-vector product  directly, but this takes a hefty
directly, but this takes a hefty  operations.
operations. . In the FFT, we needed to rearrange the columns of the DFT matrix
. In the FFT, we needed to rearrange the columns of the DFT matrix  and
and  of length
of length  and
and  respectively so that our matrix vector product can be written as
respectively so that our matrix vector product can be written as
 which takes time
which takes time  in total. Next, we compute each component
in total. Next, we compute each component  by using the formula
by using the formula
 operations to compute all the
operations to compute all the  ‘s. This leads to a total operation count of
‘s. This leads to a total operation count of  for computing the matrix-vector product
for computing the matrix-vector product  and
and  matrices
matrices  . The algorithm we presented is equivalent to evaluating this matrix triple product in the order
. The algorithm we presented is equivalent to evaluating this matrix triple product in the order  . This shows that this algorithm could be further accelerated using
. This shows that this algorithm could be further accelerated using 
 operations, just like the FFT.
operations, just like the FFT.
 . Let us by considering a special case of a Toeplitz matrix, a
. Let us by considering a special case of a Toeplitz matrix, a 
 is given by
is given by
 where
where  . This gives a fast algorithm to compute
. This gives a fast algorithm to compute  and multiply them together entrywise, take the inverse Fourier transform, and scale by
and multiply them together entrywise, take the inverse Fourier transform, and scale by  .
. of two functions
of two functions  on the real line. It is an important fact that
on the real line. It is an important fact that  (up to a possible normalizing constant).
(up to a possible normalizing constant).
 of the Toeplitz matrix Eq. (15) by
of the Toeplitz matrix Eq. (15) by  .
.  exactly equal to a power of two. This is useful because it allows us to compute matrix-vector products
exactly equal to a power of two. This is useful because it allows us to compute matrix-vector products  with the power-of-two FFT described above, which we know is fast.
with the power-of-two FFT described above, which we know is fast. by padding with zeros to get
by padding with zeros to get
 to denote matrix or vector entries which are immaterial to us. We compute
to denote matrix or vector entries which are immaterial to us. We compute  by using our fast algorithm to compute
by using our fast algorithm to compute  and then discarding everything but the first entries of
and then discarding everything but the first entries of  th entry
th entry  . We now employ a clever trick. Let
. We now employ a clever trick. Let  . Then, defining
. Then, defining  , we have that
, we have that  , which means
, which means  , where the product
, where the product  , down from proportional to
, down from proportional to  for direct evaluation. The FFT is an example of a broader matrix algorithm design strategy of looking for patterns in the numbers in a matrix and exploiting these patterns to reduce computation. The FFT can often have surprising applications, such as allowing for rapid computations with Toeplitz matrices.
for direct evaluation. The FFT is an example of a broader matrix algorithm design strategy of looking for patterns in the numbers in a matrix and exploiting these patterns to reduce computation. The FFT can often have surprising applications, such as allowing for rapid computations with Toeplitz matrices. where
where  of the set of all possible solutions
of the set of all possible solutions  . If this subspace has a basis
. If this subspace has a basis  , then the solution
, then the solution  can be represented as
can be represented as  and one only has to store the
and one only has to store the  numbers
numbers  . In general,
. In general,  .
. where
where  is the
is the  . Note that the
. Note that the  , where
, where  is the vector with zeros in all entries except for the
is the vector with zeros in all entries except for the  , we get
, we get  for all
for all  . We refer to this as a variational formulation of the linear system of equations
. We refer to this as a variational formulation of the linear system of equations 
 to the system of equations
to the system of equations 
 can be written as
can be written as  for some
for some  . Thus, writing
. Thus, writing  , we have that
, we have that  for every
for every  . But this is just a variational formulation of the equation
. But this is just a variational formulation of the equation  . The matrix
. The matrix  is SPD since
is SPD since  for
for  since
since  . Thus
. Thus  is the unique solution to the variational problem Eq. (2).
is the unique solution to the variational problem Eq. (2). and the associated
and the associated  .
. where
where  is an eigenvector of
is an eigenvector of  (
( ), then we have
), then we have  .
. satisfies all the axioms for an
satisfies all the axioms for an  between
between  for all
for all  . This follows from the straightforward calculation, for
. This follows from the straightforward calculation, for 
 since
since  since
since  to
to  is
is 
 for every
for every 
 is small
is small is a real-valued function on an interval which take to be
is a real-valued function on an interval which take to be ![[0,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-76562478d920e62fd22b998039d40967_l3.png "Rendered by QuickLaTeX.com") .
. with, for example, a
with, for example, a  .
. on the boundary
on the boundary  of the region
of the region  .
. on
on  is the
is the 
 represents, say, the temperature at a point
represents, say, the temperature at a point  measured by our thermometer will be the average temperature in the region
measured by our thermometer will be the average temperature in the region  where
where  is a weighting function which is zero outside the region
is a weighting function which is zero outside the region 
 . (The precise meaning of every will be forthcoming.) It will benefit us greatly to make this expression more “symmetric” with respect to
. (The precise meaning of every will be forthcoming.) It will benefit us greatly to make this expression more “symmetric” with respect to  . Rearranging and integrating, we see that
. Rearranging and integrating, we see that  . We then apply the
. We then apply the  , where
, where  represents integration on the surface
represents integration on the surface  for all nice functions
for all nice functions  on
on 
 , then the second two terms vanish and we’re left with the variational equation
, then the second two terms vanish and we’re left with the variational equation![\begin{equation*} \int_0^1 v'(x)u'(x) \, dx = \int_0^1 v(x) f(x) \, dx \mbox{ for all \textit{nice} functions $v$ on $[0,1]$ with } v(0) = v(1) = 0. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-38734ea04929f9afd16c7b18778f7e7d_l3.png "Rendered by QuickLaTeX.com")
 is replaced by the integral
is replaced by the integral  . The product
. The product  is replaced by the integral
is replaced by the integral  . As we shall soon see, there is a unifying theory which treats both of these contexts simultaneously.
. As we shall soon see, there is a unifying theory which treats both of these contexts simultaneously. is finite), (B) possess a square integrable derivative
is finite), (B) possess a square integrable derivative (
( is finite), and (C) are zero on the boundary. We refer to this class of functions as the
is finite), and (C) are zero on the boundary. We refer to this class of functions as the  .
. . For an arbitrary function in
. For an arbitrary function in  and
and  for an arbitrary domain
for an arbitrary domain  does not exist. Our only requirement for the variational problem is that
does not exist. Our only requirement for the variational problem is that  one has
one has  . Similarly, if one defines
. Similarly, if one defines  , then
, then  .
. is defined upon. In the linear system of equations context, this space
is defined upon. In the linear system of equations context, this space ![u : [0,1] \to \mathbb{R}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-58a33558cd429b40f98c11073bdf0af0_l3.png "Rendered by QuickLaTeX.com") as a vector of infinite length, with one entry for each real number
as a vector of infinite length, with one entry for each real number ![x \in [0,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7d5d648a174b7842da5ab1311635b25c_l3.png "Rendered by QuickLaTeX.com") .
. . We shall assume that
. We shall assume that  ) where every
) where every  of
of  smooth
smooth with the inner product
with the inner product  , the completion is
, the completion is  which we will need to establish convergence to a vector
which we will need to establish convergence to a vector  . (Think of
. (Think of  as a sequence of Galerkin approximations to a solution
as a sequence of Galerkin approximations to a solution 
 is a
is a  ). There is a beautiful and general theorem called the
). There is a beautiful and general theorem called the  ) There exists a constant
) There exists a constant  such that every
such that every  ,
,  .
. such that
such that  for every
for every  such that
such that  for every
for every  .
. for all
for all  , is no more than so much bigger or smaller than the standard inner product
, is no more than so much bigger or smaller than the standard inner product  of
of  . so the norms
. so the norms  and
and  define the same topology.
define the same topology. with inner product
with inner product  ,
,  , and
, and  . Note that we have the inequality
. Note that we have the inequality  .
. with eigenvalues
with eigenvalues  . Then
. Then  and
and  . The inequalities follow from noting the Parseval relation
. The inequalities follow from noting the Parseval relation  and noting that
and noting that  is a
is a  . Property (1) then follows from the
. Property (1) then follows from the  . Property (2) is simply the established inequality
. Property (2) is simply the established inequality  . Property (3) also follows from the Cauchy-Schwarz inequality:
. Property (3) also follows from the Cauchy-Schwarz inequality:  . Thus, by Lax-Milgram, the variational problem
. Thus, by Lax-Milgram, the variational problem  for any positive constant
for any positive constant  -inner product is given by
-inner product is given by  ,
,  . Condition (1) is follows from a application of the
. Condition (1) is follows from a application of the  . Second, we apply the Cauchy-Schwarz inequality for the vectors
. Second, we apply the Cauchy-Schwarz inequality for the vectors  and
and  to get
to get  where, e.g.,
where, e.g.,  is the Euclidean norm of the vector
is the Euclidean norm of the vector  . Next, we apply the
. Next, we apply the  . Finally, we note that
. Finally, we note that  and thus obtain
and thus obtain  . This is the desired inequality with constant one.
. This is the desired inequality with constant one.
 . This establishes Property (1) with constant
. This establishes Property (1) with constant  . As we already see one third of the way into verifying the hypotheses of Lax-Milgram, establishing these inequalities can require several steps. Ultimately, however, strong knowledge of just a core few inequalities (e.g. Cauchy-Schwarz) may be all that’s needed.
. As we already see one third of the way into verifying the hypotheses of Lax-Milgram, establishing these inequalities can require several steps. Ultimately, however, strong knowledge of just a core few inequalities (e.g. Cauchy-Schwarz) may be all that’s needed. for a constant
for a constant  ,
, . Applying the Cauchy-Schwarz inequality for integrals gives
. Applying the Cauchy-Schwarz inequality for integrals gives  since
since  . Thus
. Thus  for all
for all  . This proves Poincaré’s inequality with the constant
. This proves Poincaré’s inequality with the constant  .
.

 .
.
 to the problem Eq. (13). Moreover, the special property of Galerkin approximation holds: the error
to the problem Eq. (13). Moreover, the special property of Galerkin approximation holds: the error  is
is  ,
, 
 and Eq. (13) for
and Eq. (13) for  . Note the similarities with Eq. (3). Thus, using the Pythagorean theorem for the
. Note the similarities with Eq. (3). Thus, using the Pythagorean theorem for the  , we have
, we have
 and
and  of the best approximation in the norm
of the best approximation in the norm  . This is known as
. This is known as  be a
be a  . Since
. Since  form a basis of
form a basis of  .
. can be written as
can be written as  , so
, so  .
. and
and  into Eq. (13), we get (using bilinearity of
into Eq. (13), we get (using bilinearity of 
 and
and  , then this gives us a matrix equation
, then this gives us a matrix equation  parametrizing
parametrizing  and
and  aren’t nonzero at the same point
aren’t nonzero at the same point  . A matrix that is not sparse is said to be dense.
. A matrix that is not sparse is said to be dense. of a matrix
of a matrix  space rather than the standard
space rather than the standard  for an
for an  . Initialize the vector
. Initialize the vector  to
to  time since it requires
time since it requires  operations to initialize the vector
operations to initialize the vector  , which is true of most sparse matrices occurring in practice
, which is true of most sparse matrices occurring in practice (or matrix-tranpose-vector products
(or matrix-tranpose-vector products  ).
). . A counterexample is
. A counterexample is
 . We have that
. We have that  but
but
 nonzero elements and requires
nonzero elements and requires  and the multiplication can be done in
and the multiplication can be done in  entry of
entry of 
 ‘s denote nonzero entries and blanks denote zero entries. We then repeat the process on the
‘s denote nonzero entries and blanks denote zero entries. We then repeat the process on the  submatrix in the bottom right (the so-called
submatrix in the bottom right (the so-called 
 for
for  is a
is a  for
for  or
or  for
for  . These different forms all contain the same basic information, so we shall stick with the
. These different forms all contain the same basic information, so we shall stick with the  formulation in this post.
formulation in this post.

 ) not in the original sparse matrix
) not in the original sparse matrix 
 .
. instead. In fact, the entire
instead. In fact, the entire 
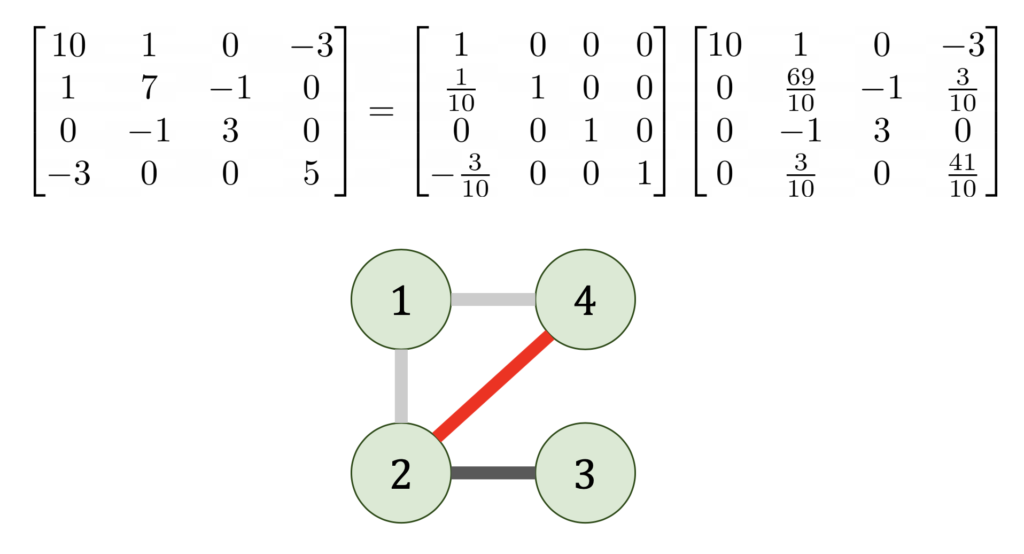
 where
where  is the Cholesky factorization of
is the Cholesky factorization of  whose nodes are the indices
whose nodes are the indices  if, and only if,
if, and only if,  .
.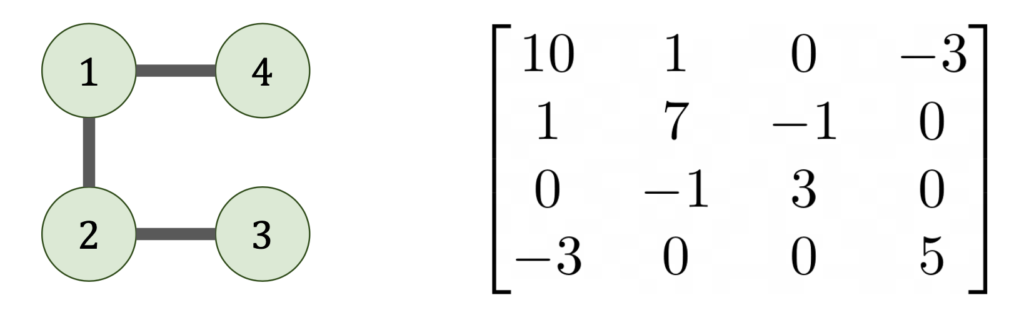
 , that finding the best elimination ordering would be a hard computational problem than the worst-case
, that finding the best elimination ordering would be a hard computational problem than the worst-case  complexity for doing Gaussian elimination in any ordering! One should not be too pessimistic about this result, however, since (assuming
complexity for doing Gaussian elimination in any ordering! One should not be too pessimistic about this result, however, since (assuming  nonzero entries. Thus, our proposition cannot hold in unmodified form.
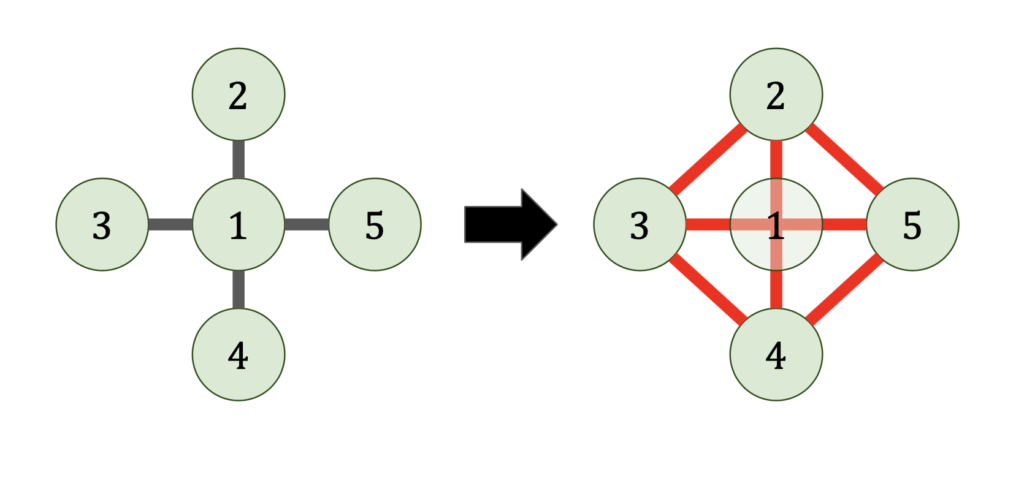
nonzero entries. Thus, our proposition cannot hold in unmodified form. 2D grid graph.
2D grid graph.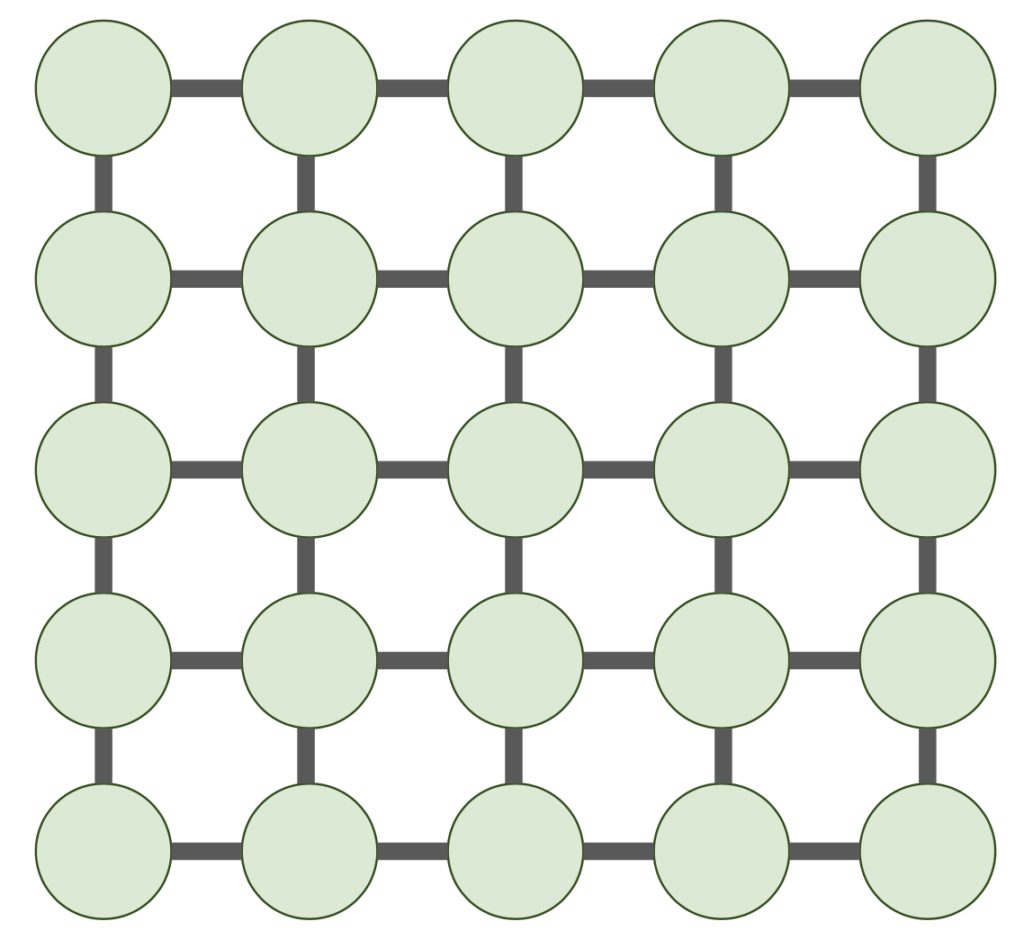
 nonzero entries. Unforunately, no linear time elimination ordering exists. We have the following theorem:
nonzero entries. Unforunately, no linear time elimination ordering exists. We have the following theorem: 2D grid graph, in any elimination ordering, the Cholesky factorization
2D grid graph, in any elimination ordering, the Cholesky factorization  requires
requires  operations and satisfies
operations and satisfies  .
. as “
as “ is no less than a constant multiple of
is no less than a constant multiple of  , asymptotically”.
, asymptotically”. -dimensional grid graphs give bounds of
-dimensional grid graphs give bounds of  time and
time and  for
for  . In particular, for 2D finite difference and finite element discretizations, sparse Cholesky factorization takes
. In particular, for 2D finite difference and finite element discretizations, sparse Cholesky factorization takes  operations and produces a Cholesky factor with
operations and produces a Cholesky factor with  in the best possible ordering.
in the best possible ordering. operations and with
operations and with  nonzero entries in the Cholesky factor.
nonzero entries in the Cholesky factor. if
if  and
and  .
. and there are no edges between
and there are no edges between  and
and  for
for  individually.
individually. dividing the graph into
dividing the graph into  roughly
roughly  pieces.
pieces.  denote the number of nonzeros in
denote the number of nonzeros in  2D grid graphs. After doing this, for step 3, all of the vertices of the separator might be connected to each other, so the separator graph will potentially have as many as
2D grid graphs. After doing this, for step 3, all of the vertices of the separator might be connected to each other, so the separator graph will potentially have as many as  edges, which result in nonzero entries in
edges, which result in nonzero entries in 
 . If one instead wants the time
. If one instead wants the time  required to compute the Cholesky factorization, note that for step 3, in the worst case, all of the vertices of the separator might be connected to each other, leading to a
required to compute the Cholesky factorization, note that for step 3, in the worst case, all of the vertices of the separator might be connected to each other, leading to a  , we get the recurrence
, we get the recurrence
 .
. , respectively. For truly large-scale problems, these limitations may be prohibitive for using such methods.
, respectively. For truly large-scale problems, these limitations may be prohibitive for using such methods. of degree
of degree  and then store the
and then store the  . From this list of numbers, we then can reconstitute an approximate version of the function. For instance, in our polynomial example, our approximate version of the function is just
. From this list of numbers, we then can reconstitute an approximate version of the function. For instance, in our polynomial example, our approximate version of the function is just  . This basic result appears in many variants in approximation theory with different precise definitions of the term “
. This basic result appears in many variants in approximation theory with different precise definitions of the term “ -period function
-period function  is a function defined on
is a function defined on ![[-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-634fe4705a526f221e312cffd3ceda93_l3.png "Rendered by QuickLaTeX.com") by
by  . This linear rescaling of the domain is very simple and easy to understand. Now define a
. This linear rescaling of the domain is very simple and easy to understand. Now define a  by
by  . There are very close connections between approximation of
. There are very close connections between approximation of 
 sense
sense , where
, where  is the truncated Fourier series
is the truncated Fourier series  and
and  .
.
 terms acts as an arbitrarily close approximate of
terms acts as an arbitrarily close approximate of  of terms we need to store might be quite large if a large value of
of terms we need to store might be quite large if a large value of  to be small. However, as we shall soon see, if the function
to be small. However, as we shall soon see, if the function  will be small for even moderate values of
will be small for even moderate values of  exists for every integer
exists for every integer  . In this post, we shall speak of smoothness as a more graded notion: loosely, a function
. In this post, we shall speak of smoothness as a more graded notion: loosely, a function 
 , as computed with the Plancherel theorem, is finite:
, as computed with the Plancherel theorem, is finite:  . This “derivative”
. This “derivative”  possesses a derivative
possesses a derivative  for
for  and
and  for
for  . For the derivative of
. For the derivative of  for
for  , then we say that
, then we say that  . The Sobolev space
. The Sobolev space 
 is a quantitative measure of qualitative smoothness. The smaller the Sobolev norm
is a quantitative measure of qualitative smoothness. The smaller the Sobolev norm 
 (that is,
(that is, 
 ” follows from the fact that
” follows from the fact that  for
for  . This very important result is a precise instantiation of our rule of thumb from earlier: if
. This very important result is a precise instantiation of our rule of thumb from earlier: if  for every
for every 
 or
or  , but for more modern problems in data science, we might be interested in very large dimensions
, but for more modern problems in data science, we might be interested in very large dimensions  denote the set of all
denote the set of all ![\begin{equation*} f(\theta) = \sum_{k \in \mathbb{Z}^d} f_{\hat k} e^{{\rm i}k\cdot \theta}, \quad \hat{f}_k = \frac{1}{(2\pi)^d} \int_{[-\pi,\pi]^d} f(\theta) e^{-{\rm i}k\cdot \theta} \, d\theta. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-530a6039fe465731c7f5274fc8e78e5e_l3.png "Rendered by QuickLaTeX.com")
 to be the
to be the  . A natural generalization of the Plancherel theorem holds as well. Let
. A natural generalization of the Plancherel theorem holds as well. Let  denote the maximum of
denote the maximum of  . Then, we have the truncated Fourier series
. Then, we have the truncated Fourier series  . Using the same calculations from the previous section, we deduce a very similar approximation property
. Using the same calculations from the previous section, we deduce a very similar approximation property
 terms! In order to include each of the first
terms! In order to include each of the first 
 is a constant depending only on
is a constant depending only on  .
. ?
? with
with  ! Consequently, there is a one-parameter approximation procedure that approximates every function perfectly. The problem with this one-parameter approximation is obvious: the one-parameter approximation is terrible at approximating most functions different than
! Consequently, there is a one-parameter approximation procedure that approximates every function perfectly. The problem with this one-parameter approximation is obvious: the one-parameter approximation is terrible at approximating most functions different than  . Call this class
. Call this class  which takes
which takes  to a tuple
to a tuple  followed by a function
followed by a function  which takes
which takes  and outputs a continuous
and outputs a continuous  .
.
 is a continuous function where
is a continuous function where  . Equivalently, the worst-case approximation error for a function in
. Equivalently, the worst-case approximation error for a function in  ,
,  .
. , one can check by carrying out the multiplication that the following factorization holds:
, one can check by carrying out the multiplication that the following factorization holds:
 denote an identity matrix of size
denote an identity matrix of size  and
and  the
the  zero matrix. Here, we use the notation of
zero matrix. Here, we use the notation of  matrix is written out as a
matrix is written out as a  “block” matrix whose entries themselves are matrices of the appropriate size that all matrices occurring in one block row (or column) have the same number of rows (or columns). Two block matrices which are blocked in a compatible way can be multiplied just like two regular matrices can be multiplied, taking care of the noncommutativity of matrix multiplication.
“block” matrix whose entries themselves are matrices of the appropriate size that all matrices occurring in one block row (or column) have the same number of rows (or columns). Two block matrices which are blocked in a compatible way can be multiplied just like two regular matrices can be multiplied, taking care of the noncommutativity of matrix multiplication. . One can verify that for a block triangular matrix
. One can verify that for a block triangular matrix  , we have the inverse formula
, we have the inverse formula
 for the proposed formula for
for the proposed formula for  and verifying that one obtains the identity matrix.) A similar formula holds for block lower triangular matrices. From here, we can deduce a formula for the inverse of
and verifying that one obtains the identity matrix.) A similar formula holds for block lower triangular matrices. From here, we can deduce a formula for the inverse of  be the Schur complement. Then
be the Schur complement. Then
 ,
,  ,
,  -block entry of
-block entry of  is simply just the inverse of the Schur complement.
is simply just the inverse of the Schur complement. . Notice that this is the Schur complement of the matrix
. Notice that this is the Schur complement of the matrix  , which is the same
, which is the same  such that
such that  . Alternately, and perhaps more cleanly, one may define two Schur complements of the block matrix
. Alternately, and perhaps more cleanly, one may define two Schur complements of the block matrix  : one by “eliminating
: one by “eliminating  , and the other by “eliminating
, and the other by “eliminating  .
. appears in the
appears in the  , the inverse of the alternate Schur complement
, the inverse of the alternate Schur complement 
 . Using the Sherman-Morrison-Woodbury identity with
. Using the Sherman-Morrison-Woodbury identity with  ,
,  , and
, and  , we have that
, we have that
 for
for  and
and  for
for  .
. where
where 
 is the Schur complement of the matrix
is the Schur complement of the matrix  , where
, where  . It is well known that the solution
. It is well known that the solution  . However, if the matrix
. However, if the matrix  will be much more ill-conditioned (
will be much more ill-conditioned (
 . Thus, we can solve the normal equations by instead solving the (potentially, see footnote) much better-conditioned system
. Thus, we can solve the normal equations by instead solving the (potentially, see footnote) much better-conditioned system to be proportional to the smallest
to be proportional to the smallest  of
of  , then one can show that the two-norm condition number of
, then one can show that the two-norm condition number of  , then the two-norm condition number of
, then the two-norm condition number of 
 which simplifies to
which simplifies to  . The unknown
. The unknown  , which encodes the condition that the residual is orthogonal to the range of the matrix
, which encodes the condition that the residual is orthogonal to the range of the matrix