In this post, I want to discuss a very beautiful piece of mathematics I stumbled upon recently. As a warning, this post will be more mathematical than most, but I will still try and sand off the roughest mathematical edges. This post is adapted from a much more comprehensive post by Paata Ivanishvili. My goal is to distill the main idea to its essence, deferring the stochastic calculus until it cannot be avoided.
Jensen’s inequality is one of the most important results in probability.
Jensen’s inequality. Let be a (real) random variable and a convex function such that both and are defined. Then .
Here is the standard proof. A convex function has supporting lines. That is, at a point , there exists a slope such that for all . Invoke this result at and and take expectations to conclude that
In this post, I will outline a proof of Jensen’s inequality which is much longer and more complicated. Why do this? This more difficult proof illustrates an incredible powerful technique for proving inequalities, interpolation. The interpolation method can be used to prove a number of difficult and useful inequalities in probability theory and beyond. As an example, at the end of this post, we will see the Gaussian Jensen inequality, a striking generalization of Jensen’s inequality with many applications.
The idea of interpolation is as follows: Suppose I wish to prove for two numbers and . This may hard to do directly. With the interpolation method, I first construct a family of numbers , , such that and and show that is (weakly) increasing in . This is typically accomplished by showing the derivative is nonnegative:
To prove Jensen’s inequality by interpolation, we shall begin with a special case. As often in probability, the simplest case is that of a Gaussian random variable.
Jensen’s inequality for a Gaussian. Let be a standard Gaussian random variable (i.e., mean-zero and variance ) and let be a thrice-differentiable convex function satisfying a certain technical condition.1Specifically, we assume the regularity condition for some for any Gaussian random variable . Then
Note that the conclusion is exactly Jensen’s inequality, as we have assumed is mean-zero.
The difficulty with any proof by interpolation is to come up with the “right” . For us, the “right” answer will take the form
where starts with no randomness and is our standard Gaussian. To interpolate between these extremes, we increase the variance linearly from to . Thus, we define
Here, and throughout, denotes a Gaussian random variable with zero mean and variance .
Let’s compute the derivative of . To do this, let denote a small parameter which we will later send to zero. For us, the key fact will be that a can be realized as a sum of independent and random variables. Therefore, we write
We now evaluate by using Taylor’s formula
(1)
where lies between and . Now, take expectations,
The random variable has mean zero and variance so this gives
As we show below, the remainder term vanishes as . Thus, we can rearrange this expression to compute the derivative:
Jensen’s inequality is proven! In fact, we’ve proven the stronger version of Jensen’s inequality:
This strengthened version can yield improvements. For instance, if is -smooth
then we have
This inequality isn’t too hard to prove directly, but it does show that we’ve obtained something more than the simple proof of Jensen’s inequality.
Analyzing the Remainder Term
Let us quickly check that the remainder term vanishes as . Let’s do this. As an exercise, you can verify that our technical regularity condition implies . Thus, by Hölder’s inequality and setting to be ‘s Hölder conjugate (), we obtain
One can show that where is a function of alone. Therefore, as .
What’s Really Going On Here?
In our proof, we use a family of random variables , defined for each . Rather than treating these quantities as independent, we can think of them as a collective, comprising a random function known as a Brownian motion.
The Brownian motion is a very natural way of interpolating between a constant and a Gaussian with mean .2The Ornstein–Uhlenbeck process is another natural way of interpolating between a random variable and a Gaussian.
There is an entire subject known as stochastic calculus which allows us to perform computations with Brownian motion and other random processes. The rules of stochastic calculus can seem bizarre at first. For a function of a real number , we often write
For a function of a Brownian motion, the analog is Itô’s formula
While this might seem odd at first, this formula may seem more sensible if we compare with (1) above. The idea, very roughly, is that for an increment of the Brownian motion over a time interval , is a random variable with mean , so we cannot drop the second term in the Taylor series, even up to first order in . Fully diving into the subtleties of stochastic calculus is far beyond the scope of this short post. Hopefully, the rest of this post, which outlines some extensions of our proof of Jensen’s inequality that require more stochastic calculus, will serve as an enticement to learn more about this beautiful subject.
Proving Jensen by Interpolation
For the rest of this post, we will be less careful with mathematical technicalities. We can use the same idea that we used to prove Jensen’s inequality for a Gaussian random variable to prove Jensen’s inequality for any random variable :
Here is the idea of the proof.
First, realize that we can write any random variable as a function of a standard Gaussian random variable . Indeed, letting and denote the cumulative distribution functions of and , one can show that
has the same distribution as .
Now, as before, we can interpolate between and using a Brownian motion. As a first, idea, we might try
Unfortunately, this choice of does not work! Indeed, does not even equal to ! Instead, we must define
We define using the conditional expectation of the final value conditional on the Brownian motion at an earlier time . Using a bit of elbow grease and stochastic calculus, one can show that
This provides a proof of Jensen’s inequality in general by the method if interpolation.
Gaussian Jensen Inequality
Now, we’ve come to the real treat, the Gaussian Jensen inequality. In the last section, we saw the sketch of a proof of Jensen’s inequality using interpolation. While it is cool that this proof is possible, we learned anything new since we can prove Jensen’s inequality in other ways. The Gaussian Jensen inequality provides an application of this technique which is hard to prove other ways. This section, in particular, is cribbing quite heavily from Paata Ivanishvili‘s excellent post on the topic.
Here’s the big question:
If are “somewhat dependent”, for which functions does the multivariate Jensen’s inequality
()
hold?
Considering extreme cases, if are entirely dependent, then we would only expect () to hold when is convex. But if are independent, then we can apply Jensen’s inequality to each coordinate one at a time to deduce
We would like a result which interpolates between extremes {fully dependent, fully convex} and {independent, separately convex}. The Gaussian Jensen inequality provides exactly this tool.
As in the previous section, we can generate arbitrary random variables as functions of Gaussian random variables . We will use the covariance matrix of the Gaussian random variables as our measure of the dependence of the random variables . With this preparation in place, we have the following result:
Gaussian Jensen inequality. The conclusion of Jensen’s inequality
This is a beautiful result with striking consequences (see Ivanishvili‘s post). The proof is essentially the same as the proof as Jensen’s inequality by interpolation with a little additional bookkeeping.
Let us confirm this result respects our extreme cases. In the case where are equal (and variance one), is a matrix of all ones and for all . Thus, the Gaussian Jensen inequality states that (2) holds if and only if is positive semidefinite for every , which occurs precisely when is convex.
Next, suppose that are independent and variance one, then is the identity matrix and
A diagonal matrix is positive semidefinite if and only if its entries are nonnegative. Thus, (2) holds if and only if each of ‘s diagonal second derivatives are nonnegative : this is precisely the condition for to be separately convex in each argument.
There’s much more to be said about the Gaussian Jensen inequality, and I encourage you to read Ivanishvili‘s post to see the proof and applications. What I find so compelling about this result—so compelling that I felt the need to write this post—is how interpolation and stochastic calculus can be used to prove inequalities which don’t feel like stochastic calculus problems. The Gaussian Jensen inequality is a statement about functions of dependent Gaussian random variables; there’s nothing dynamic happening. Yet, to prove this result, we inject dynamics into the problem, viewing the two sides of our inequality as endpoints of a random process connecting them. This is a such a beautiful idea that I couldn’t help but share it.
Our paper presents new trace estimators XTrace and XNysTrace which are highly efficient, producing accurate trace approximations using a small budget of matrix–vector products. In addition, these algorithms are fast to run and are supported by theoretical results which explain their excellent performance. I really hope that you will check out the paper to learn more about these estimators!
For the rest of this post, I’m going to talk about the most basic stochastic trace estimation algorithm, the Girard–Hutchinson estimator. This seemingly simple algorithm exhibits a number of nuances and forms the backbone for more sophisticated trace estimates such as Hutch++, Nyström++, XTrace, and XNysTrace. Toward the end, this blog post will be fairly mathematical, but I hope that the beginning will be fairly accessible to all.
Girard–Hutchinson Estimator: The Basics
The Girard–Hutchinson estimator for the trace of a square matrix is
(1)
Here, are random vectors, usually chosen to be statistically independent, and denotes the conjugate transpose of a vector or matrix. The Girard–Hutchinson estimator only depends on the matrix through the matrix–vector products .
Therefore, to prove that , it is sufficient to prove that for each .
When working with traces, there are two tricks that solve 90% of derivations. The first trick is that, if we view a number as a matrix, then a number equals its trace, . The second trick is the cyclic property: For a matrix and a matrix , we have . The cyclic property should be handled with care when one works with a product of three or more matrices. For instance, we have
However,
One should think of the matrix product as beads on a closed loop of string. One can move the last bead to the front of the other two, , but not interchange two beads, .
With this trick in hand, let’s return to proving that for every . Apply our two tricks:
The expectation is a linear operation and the matrix is non-random, so we can bring the expectation into the trace as
Invoke the isotropy condition (2) and conclude:
Plugging this into (4) confirms the unbiasedness claim (3).
Variance
Continue to assume that the ‘s are isotropic (3) and now assume that are independent. By independence, the variance can be written as
Assuming that are identically distributed, we then get
The variance decreases like , which is characteristic of Monte Carlo-type algorithms. Since is unbiased (i.e, (3)), this means that the mean square error decays like so the average error (more precisely root-mean-square error) decays like
This type of convergence is very slow. If I want to decrease the error by a factor of , I must do the work!
Variance-reduced trace estimators like Hutch++ and our new trace estimator XTrace improve the rate of convergence substantially. Even in the worst case, Hutch++ and XTrace reduce the variance at a rate and (root-mean-square) error at rates :
For matrices with rapidly decreasing singular values, the variance and error can decrease much faster than this.
Variance Formulas
As the rate of convergence for the Girard–Hutchinson estimator is so slow, it is imperative to pick a distribution on test vectors that makes the variance of the single–sample estimate as low as possible. In this section, we will provide several explicit formulas for the variance of the Girard–Hutchinson estimator. Derivations of these formulas will appear at the end of this post. These variance formulas help illuminate the benefits and drawbacks of different test vector distributions.
To express the formulas, we will need some notation. For a complex number we use and to denote the real and imaginary parts. The variance of a random complex number is
We first focus on real-valued test vectors . Since is real, we can use the ordinary transpose rather than the conjugate transpose . Since is a number, it is equal to its own transpose:
Therefore,
The Girard–Hutchinson trace estimator applied to is the same as the Girard–Hutchinson estimator applied to the symmetric part of , .
Uniform signs (Rademachers): are independent random vectors with uniform coordinates.
Real sphere: Assume are uniformly distributed on the real sphere of radius : .
These formulas continue to hold for nonsymmetric by replacing by its symmetric part on the right-hand sides of these variance formulas.
Complex-Valued Test Vectors
We now move our focus to complex-valued test vectors . As a rule of thumb, one should typically expect that the variance for complex-valued test vectors applied to a real symmetric matrix is about half the natural real counterpart—e.g., for complex Gaussians, you get about half the variance than with real Gaussians.
denote the Hermitian and skew-Hermitian parts of . Similar to how the imaginary part of a complex number is real, the skew-Hermitian part of a complex matrix is Hermitian (and is skew-Hermitian). Since and are both Hermitian, we have
Consequently, the variance of can be broken into Hermitian and skew-Hermitian parts:
For this reason, we will state the variance formulas only for Hermitian , with the formula for general following from the Cartesian decomposition.
For the following results, assume is Hermitian, .
Complex Gaussian: are independent standard complex random vectors, i.e., each has iid entries distributed as for standard normal random variables.
Uniform phases (Steinhauses): are independent random vectors whose entries are uniform on the complex unit circle .
Complex sphere: Assume are uniformly distributed on the complex sphere of radius : .
Optimality Properties
Let us finally address the question of what the best choice of test vectors is for the Girard–Hutchinson estimator. We will state two results with different restrictions on .
Our first result, due to Hutchinson, is valid for real symmetric matrices with real test vectors.
Optimality (independent test vectors with independent coordinates). If the test vectors are isotropic (2), independent from each other, and have independent entries, then for any fixed real symmetric matrix , the minimum variance for is obtained when are populated with random signs .
The next optimality results will have real and complex versions. To present the results for -valued and an -valued test vectors on unified footing, let denote either or . We let a -Hermitian matrix be either a real symmetric matrix (if ) or a complex Hermitian matrix (if ). Let a -unitary matrix be either a real orthogonal matrix (if ) or a complex unitary matrix (if ).
The condition that the vectors have independent entries is often too restrictive in practice. It rules out, for instance, the case of uniform vectors on the sphere. If we relax this condition, we get a different optimal distribution:
Optimality (independent test vectors). Consider any set of -Hermitian matrices which is invariant under -unitary similary transformations:
Assume that the test vectors are independent and isotropic (2). The worst-case variance is minimized by choosing uniformly on the -sphere: .
More simply, if you wants your stochastic trace estimator to be effective for a class of inputs (closed under -unitary similarity transformations) rather than a single input matrix , then the best distribution are test vectors drawn uniformly from the sphere. Examples of classes of matrices include:
Fixed eigenvalues. For fixed real eigenvalues , the set of all -Hermitian matrices with these eigenvalues.
Frobenius norm ball. The class of all -Hermitian matrices of Frobenius norm at most 1.
Derivation of Formulas
In this section, we provide derivations of the variance formulas. I have chosen to focus on derivations which are shorter but use more advanced techniques rather than derivations which are longer but use fewer tricks.
Real Gaussians
First assume is real. Since is real symmetric, has an eigenvalue decomposition, where is orthogonal and is a diagonal matrix reporting ‘s eigenvalues. Since the real Gaussian distribution is invariant under orthogonal transformations, has the same distribution as . Therefore,
For non-real matrix, we can break the matrix into its entrywise real and imaginary parts . Thus,
Uniform Signs
First, compute
For a vector of uniform random signs, we have for every , so the second sum vanishes. Note that we have assumed symmetric, so the sum over can be replaced by two times the sum over :
Note that are pairwise independent. As a simple exercise, one can verify that the identity
holds for any pairwise independent family of random variances and numbers . Ergo,
In the second-to-last line, we use the fact that is a uniform random sign, which has variance . The final line is a consequence of the symmetry of .
Uniform on the Real Sphere
The simplest proof is I know is by the “camel principle”. Here’s the story (a lightly edited quotation from MathOverflow):
A father left 17 camels to his three sons and, according to the will, the eldest son was to be given a half of the camels, the middle son one-third, and the youngest son the one-ninth. The sons did not know what to do since 17 is not evenly divisible into either two, three, or nine parts, but a wise man helped the sons: he added his own camel, the oldest son took camels, the second son took camels, the third son camels and the wise man took his own camel and went away.
We are interested in a vector which is uniform on the sphere of radius . Performing averages on the sphere is hard, so we add a camel to the problem by “upgrading” to a spherically symmetric vector which has a random length. We want to pick a distribution for which the computation is easy. Fortunately, we already know such a distribution, the Gaussian distribution, for which we already calculated .
The Gaussian vector and the uniform vector on the sphere are related by
where is the squared length of the Gaussian vector . In particular, has the distribution of the sum of squared Gaussian random variables, which is known as a random variable with degrees of freedom.
Here, and denote the conditional variance and conditional expectation with respect to the random variable . The quick and dirty ways of working with these are to treat the random variable “like a constant” with respect to the conditional variance and expectation.
Plugging in the formula and treating “like a constant”, we obtain
As we mentioned, is a random variable with degrees of freedom and and are known quantities that can be looked up:
We know and . Plugging these all in, we get
Rearranging, we obtain
Complex Gaussians
The trick is the same as for real Gaussians. By invariance of complex Gaussian random vectors under unitary transformations, we can reduce to the case where is a diagonal matrix populated with eigenvalues . Then
Since is uniformly distributed on the complex unit circle, we can assume without loss of generality that . Thus, letting be uniform on the complex unit circle,
The real and imaginary parts of have the same distribution so
so . Thus
Uniform on the Complex Sphere: Derivation 1 by Reduction to Real Case
There are at least three simple ways of deriving this result: the camel trick, reduction to the real case, and Haar integration. Each of these techniques illustrates a trick that is useful in its own right beyond the context of trace estimation. Since we have already seen an example of the camel trick for the real sphere, I will present the other two derivations.
Let us begin with the reduction to the real case. Let and denote the real and imaginary parts of a vector or matrix, taken entrywise. The key insight is that if is a uniform random vector on the complex sphere of radius , then
We’ve converted the complex vector into a real vector .
Now, we need to convert the complex matrix into a real matrix . To do this, recall that one way of representing complex numbers is by matrices:
Using this correspondence addition and multiplication of complex numbers can be carried by addition and multiplication of the corresponding matrices.
To convert complex matrices to real matrices, we use a matrix-version of the same representation:
One can check that addition and multiplication of complex matrices can be carried out by addition and multiplication of the corresponding “realified” matrices, i.e.,
holds for all complex matrices and .
We’ve now converted complex matrix and vector into real matrix and vector . Let’s compare to . A short calculation reveals
Since is a uniform random vector on the sphere of radius , is a uniform random vector on the sphere of radius . Thus, by the variance formula for the real sphere, we get
A short calculation verifies that and . Plugging this in, we obtain
Uniform on the Complex Sphere: Derivation 2 by Haar Integration
The proof by reduction to the real case requires some cumbersome calculations and requires that we have already computed the variance in the real case by some other means. The method of Haar integration is more slick, but it requires some pretty high-power machinery. Haar integration may be a little bit overkill for this problem, but this technique is worth learning as it can handle some truly nasty expected value computations that appear, for example, in quantum information.
We seek to compute
The first trick will be to write this expession using a single matrix trace using the tensor (Kronecker) product. For those unfamiliar with the tensor product, the main properties we will be using are
(6)
We saw in the proof of unbiasedness that
Therefore, by (6),
Thus, to evaluate , it will be sufficient to evaluate . Forunately, there is a useful formula for these expectation provided by a field of mathematics known as representation theory (see Lemma 1 in this paper):
To evalute the trace on the right-hand side of this equation, there is another formula (see Lemma 6 in this paper):
Therefore, we conclude
Proof of Optimality Properties
In this section, we provide proofs of the two optimality properties.
Optimality: Independent Vectors with Independent Coordinates
Assume is real and symmetric and suppose that is isotropic (2) with independent coordinates. The isotropy condition
implies that , where is the Kronecker symbol. Using this fact, we compute the second moment:
Thus
The variance is minimized by choosing with as small as possible. Since , the smallest possible value for is , which is obtained by populating with random signs.
Optimality: Independent Vectors
This result appears to have first been proven by Richard Kueng in unpublished work. We use an argument suggested to me by Robert J. Webber.
Assume is a class of -Hermitian matrices closed under -unitary similarity transformations and that is an isotropic random vector (2). Decompose the test vector as
First, we shall show that the variance is reduced by replacing with a vector drawn uniformly from the sphere
(7)
where
(8)
Note that such a can be generated as for a uniformly random -unitary matrix . Therefore, we have
Let’s start with a classical problem: connect-the-dots. As we know from geometry, any two points in the plane are connected by one and only one straight line:
But what if we have more than two points? How should we connect them? One natural way is by parabola. Any three points (with distinct coordinates) are connected by one and only one parabola :
And we can keep extending this. Any points1The degree of the polynomial is one less than the number of points because a degree- polynomial is described by coefficients. For instance, a degree-two parabola has three coefficients , , and . (with distinct coordinates) are connected by a unique degree- polynomial :
This game of connect-the-dots with polynomials is known more formally as polynomial interpolation. We can use polynomial interpolation to approximate functions. For instance, we can approximate the function on the interval to visually near-perfect accuracy by connecting the dots between seven points :
But something very peculiar happens when we try and apply this trick to the specially chosen function on the interval :
Unlike , the polynomial interpolant for is terrible! What’s going on? Why doesn’t polynomial interpolation work here? Can we fix it? The answer to the last question is yes and the solution is Chebyshev polynomials.
Reverse-Engineering Chebyshev
The failure of polynomial interpolation for is known as Runge’s phenomenon after Carl Runge who discovered this curious behavior in 1901. The function is called the Runge function. Our goal is to find a fix for polynomial interpolation which crushes the Runge phenomenon, allowing us to reliably approximate every sensible2A famous theorem of Faber states that there does not exist any set of points through which the polynomial interpolants converge for every continuous function. This is not as much of a problem as it may seem. As the famous Weierstrass function shows, arbitrary continuous functions can be very weird. If we restrict ourselves to nicer functions, such as Lipschitz continuous functions, there does exist a set of points through which the polynomial interpolant always converges to the underlying function. Thus, in this senses, it is possible to crush the Runge phenomenon. function with polynomial interpolation.
Carl Runge
Let’s put on our thinking caps and see if we can discover the fix for ourselves. In order to discover a fix, we must first identify the problem. Observe that the polynomial interpolant is fine near the center of the interval; it only fails near the boundary.
This leads us to a guess for what the problem might be; maybe we need more interpolation points near the boundaries of the interval. Indeed, tipping our hand a little bit, this turns out to be the case. For instance, connecting the dots for the following set of “mystery points” clustered at the endpoints works just fine:
Let’s experiment a little and see if we can discover a nice set of interpolation points, which we will call , like this for ourselves. We’ll assume the interpolation points are given by a function so we can form the polynomial interpolant for any desired polynomial degree .3Technically, we should insist on the function being \textit{injective} so that the points are guaranteed to be distinct. For instance, if we pick , the points look like this:
Equally spaced points (shown on vertical axis) give rise to non-equally spaced points (shown on horizontal axis)
How should we pick the function ? First observe that, even for the Runge function, equally spaced interpolation points are fine near the center of the interval. We thus have at least two conditions for our desired interpolation points:
The interior points should maintain their spacing of roughly .
The points must cluster near both boundaries.
As a first attempt let’s divide the interval into thirds and halve the spacing of points except in the middle third. This leads to the function
These interpolation points initially seem promising, even successfully approximating the Runge function itself.
Unfortunately, this set of points fails when we consider other functions. For instance, if we use the Runge-like function , we see that these interpolation points now lead to a failure to approximate the function at the middle of the interval, even if we use a lot of interpolation points!
Maybe the reason this set of interpolation points didn’t work is that the points are too close at the endpoints. Or maybe we should have divided the interval as quarter–half–quarter rather than thirds. There are lots of variations of this strategy for choosing points to explore and all of them eventually lead to failure on some Runge-flavored example. We need a fundamentally different strategy then making the points times closer within distance of the endpoints.
Let’s try a different approach. The closeness of the points at the endpoints is determined by the slope of the function at and . The smaller that and are, the more clustered the points will be. For instance,
When we halved the distance between points, we instead had
So if we want the points to be much more clustered together, it is natural to require
It also makes sense for the function to cluster points equally near both endpoints, since we see no reason to preference one end over the other. Collecting together all the properties we want the function to have, we get the following list:
spans the whole range ,
, and
is symmetric about , .
Mentally scrolling through our Rolodex of friendly functions, a natural one that might come to mind meeting these three criteria is the cosine function, specifically . This function yields points which are more clustered at the endpoints:
The points
we guessed our way into are known as the Chebyshev points.4Some authors refer to these as the “Chebyshev points of the second kind” or use other names. We follow the convention in Approximation Theory and Approximation Practice (Chapter 1) and simply refer to these points simply as the Chebyshev points. The Chebyshev points prove themselves perfectly fine for the Runge function:
As we saw earlier, success on the Runge function alone is not enough to declare victory for the polynomial interpolation problem. However, in this case, there are no other bad examples left to find. For any nice function with no jumps, polynomial interpolation through the Chebyshev points works excellently.5Specifically, for a function which not too rough (i.e., Lipschitz continuous), the degree- polynomial interpolant of through the Chevyshev points converges uniformly to as .
Why the Chebyshev Points?
We’ve guessed our way into a solution to the polynomial interpolation problem, but we still really don’t know what’s going on here. Why are the Chebyshev points much better at polynomial interpolation than equally spaced ones?
Now that we know that the Chebyshev points are a right answer to the interpolation problem,6Indeed, there are other sets of interpolation points through which polynomial interpolation also works well, such as the Legendre points. let’s try and reverse engineer a principled reason for why we would expect them to be effective for this problem. To do this, we ask:
What is special about the cosine function?
From high school trigonometry, we know that gives the coordinate of a point radians along the unit circle. This means that the Chebyshev points are the coordinates of equally spaced points on the unit circle (specifically the top half of the unit circle ).
Chebyshev points are the coordinates of equally spaced points on the unit circle.
This raises the question:
What does the interpolating polynomial look like as a function of the angle?
To convert between and we simply plug in to :
This new function depending on , which we can call , is a polynomial in the variable. Powers of cosines are not something we encounter every day, so it makes sense to try and simplify things using some trig identities. Here are the first couple powers of cosines:
A pattern has appeared! The powers always take the form7As a fun exercise, you might want to try and prove this using mathematical induction.
The significance of this finding is that, by plugging in each of these formulas for , we see that our polynomial in the variable has morphed into a Fourier cosine series in the variable :
For anyone unfamiliar with Fourier series, we highly encourage the 3Blue1Brown video on the subject, which explains why Fourier series are both mathematically beautiful and practically useful. The basic idea is that almost any function can be expressed as a combination of waves (that is, sines and cosines) with different frequencies.8More precisely, we might call these angular frequencies. In our case, this formula tells us that is equal to units of frequency , plus units of frequency , all the way up to units of frequency . Different types of Fourier series are appropriate in different contexts. Since our Fourier series only possesses cosines, we call it a Fourier cosine series.
We’ve discovered something incredibly cool:
Polynomial interpolation through the Chebyshev points is equivalent to finding a Fourier cosine series for equally spaced angles .
We’ve arrived at an answer to why the Chebyshev points work well for polynomial interpolation.
Polynomial interpolation through the Chebyshev points is effective because Fourier cosine series through equally spaced angles is effective.
Of course, this explanation just raises the further question: Why do Fourier cosine series give effective interpolants through equally spaced angles ? This question has a natural answer as well, involving the convergence theory and aliasing formula (see Section 3 of this paper) for Fourier series. We’ll leave the details to the interested reader for investigation. The success of Fourier cosines series in interpolating equally spaced data is a fundamental observation that underlies the field of digital signal processing. Interpolation through the Chebyshev points effectively hijacks this useful fact and applies it to the seemingly unrelated problem of polynomial interpolation.
Another question this explanation raises is the precise meaning of “effective”. Just how good are polynomial interpolants through the Chebyshev points at approximating functions? As is discussed at length in another post on this blog, the degree to which a function can be effectively approximated is tied to how smooth or rough it is. Chebyshev interpolants approximate nice analytic functions like or with exponentially small errors in the number of interpolation points used. By contrast, functions with kinks like are approximated with errors which decay much more slowly. See theorems 2 and 3 on this webpage for more details.
Chebyshev Polynomials
We’ve now discovered a set of points, the Chebyshev points, through which polynomial interpolation works well. But how should we actually compute the interpolating polynomial
Again, it will be helpful to draw on the connection to Fourier series. Computations with Fourier series are highly accurate and can be made lightning fast using the fast Fourier transform algorithm. By comparison, directly computing with a polynomial through its coefficients is a computational nightmare.
In the variable , the interpolant takes the form
To convert back to , we use the inverse function9One always has to be careful when going from to since multiple values get mapped to a single value by the cosine function. Fortunately, we’re working with variables and , between which the cosine function is one-to-one with the inverse function being given by the arccosine. to obtain:
This is a striking formula. Given all of the trigonometric functions, it’s not even obvious that is a polynomial (it is)!
Despite its seeming peculiarity, this is a very powerful way of representing the polynomial . Rather than expressing using monomials, we’ve instead written as a combination of more exotic polynomials
The polynomials are known as the Chebyshev polynomials,10More precisely, the polynomials are known as the Chebyshev polynomials of the first kind. named after Pafnuty Chebyshev who studied the polynomials intensely.11The letter “T” is used for Chebyshev polynomials since the Russian name “Chebyshev” is often alternately transliterated to English as “Tchebychev”.
Pafnuty Chebyshev
Writing out the first few Chebyshev polynomials shows they are indeed polynomials:
Since and are both polynomials, every Chebyshev polynomial is as well.
We’ve arrived at the following amazing conclusion:
Under the change of variables , the Fourier cosine series
becomes the combination of Chebyshev polynomials
This simple and powerful observations allows us to apply the incredible speed and accuracy of Fourier series to polynomial interpolation.
Beyond being a neat idea with some nice mathematics, this connection between Fourier series and Chebyshev polynomials is a powerful tool for solving computational problems. Once we’ve accurately approximated a function by a polynomial interpolant, many quantities of interest (derivatives, integrals, zeros) become easy to compute—after all, we just have to compute them for a polynomial! We can also use Chebyshev polynomials to solve differential equations with much faster rates of convergence than other methods. Because of the connection to Fourier series, all of these computations can be done to high accuracy and blazingly fast via the fast Fourier transform, as is done in the software package Chebfun.
The Chebyshev polynomials have an array of amazing properties and they appear all over mathematics and its applications in other fields. Indeed, we have only scratched the surface of the surface. Many questions remain:
What is the connection between the Chebyshev points and the Chebyshev polynomials?
The cosine functions are orthogonal to each other; are the Chebyshev polynomials?
Are the Chebyshev points the best points for polynomial interpolation? What does “best” even mean in this context?
Every “nice” even periodic function has an infinite Fourier cosine series which converges to it. Is there a Chebyshev analog? Is there a relation between the infinite Chebyshev series and the (finite) interpolating polynomial through the Chebyshev points?
All of these questions have beautiful and fairly simple answers. The book Approximation Theory and Approximation Practice is a wonderfully written book that answers all of these questions in its first six chapters, which are freely available on the author’s website. We recommend the book highly to the curious reader.
TL;DR: To get an accurate polynomial approximation, interpolate through the Chebyshev points. To compute the resulting polynomial, change variables to , compute the Fourier cosine series interpolant, and obtain your polynomial interpolant as a combination of Chebyshev polynomials.
The (ordinary) linear least squares problem is as follows: given an matrix and a vector of length , find the vector such that is as close to as possible, when measured using the two-norm. That is, we seek to
(1)
From this equation, the name “least squares” is self-explanatory: we seek which minimizes the sum of the squared discrepancies between the entries of and .
The least squares problem is ubiquitous in science, engineering, mathematics, and statistics. If we think of each row of as an input and its corresponding entry of as an output, then the solution to the least squares model gives the coefficients of a linear model for the input–output relationship. Given a new previously unseen input , our model predicts the output is approximately . The vector consists of coefficients for this linear model. The least squares solution satisfies the property that the average squared difference between the output and the prediction is as small as it could possibly be for all choices of coefficient vectors .
How do we solve the least squares problem? A classical solution approach, ubiquitous in textbooks, is to solve a system of linear equations known as the normal equations. The normal equations associated with the least squares problem (1) are given by
(2)
This system of equations always has a solution. If has full column-rank, then is invertible and the unique least squares solution to (1) is given by . We assume that has full column-rankQ for the rest of this discussion. To solve the normal equations in software, we compute and and solve (2) using a linear solver like MATLAB’s “\”.1Even better, we could us a Cholesky decomposition since the matrix is positive definite. (As is generally true in matrix computations, it is almost never a good idea to explicitly form the inverse of the matrix , or indeed any matrix.) We also can solve the normal equations using an iterative method like (preconditioned) conjugate gradient.
The purpose of the article is to advocate against the use of the normal equations for solving the least squares problems, at least in most cases. So what’s wrong with the normal equations? The problem is not that the normal equations aren’t mathematically correct. Instead, the problem is that the normal equations often lead to poor accuracy for the least squares solution using computer arithmetic.
Most of the time when using computers, we store real numbers as floating point numbers.2One can represent rational numbers on a computer as fractions of integers and operations can be done exactly. However, this is prone to gross inefficiencies as the number of digits in the rational numbers can grow to be very large, making the storage and time to solve linear algebra problems with rationals dramatically more expensive. For these reasons, the vast majority of numerical computations use floating point numbers which store only a finite number of digits for any given real number. In this model, except for extremely rare circumstances, rounding errors during arithmetic operations are a fact of life. At a coarse level, the right model to have in your head is that real numbers on a computer are stored in scientific notation with only 16 decimal digits after the decimal point.3This is a simplification in multiple ways. First, computers store numbers in binary and thus, rather than storing 16 decimal digits after the decimal point, they store 52 binary digits. This amounts to roughly 16 decimal digits. Secondly, there are different formats for storing real numbers as floating point on a computer with different amounts of stored digits. The widely used IEEE double precision format has about 16 decimal digits of accuracy; the IEEE single precision format has roughly 8. When two numbers are added, subtracted, multiplied, and divided, the answer is computed and then rounded to 16 decimal digits; any extra digits of information are thrown away. Thus, the result of our arithmetic on a computer is the true answer to the arithmetic problem plus a small rounding error. These rounding errors are small individually, but solving an even modestly sized linear algebra problem requires thousands of such operations. Making sure many small errors don’t pile up into a big error is part of the subtle art of numerical computation.
To make a gross simplification, if one solves a system of linear equations on a computer using a well-designed piece of software, one obtains an approximate solution which is, after accounting for the accumulation of rounding errors, close to . But just how close the computed solution and the true solution are depends on how “nice” the matrix is. The “niceness” of a matrix is quantified by a quantity known as the condition number of , which we denote .4In fact, there are multiple definitions of the condition number depending on the norm which one uses the measure the sizes of vectors. Since we use the 2-norm, the appropriate 2-norm condition number is the ratio of the largest and smallest singular values of . As a rough rule of thumb, the relative error between and is roughly bounded as
(3)
The “ corresponds to the fact we have roughly 16 decimal digits of accuracy in double precision floating point arithmetic. Thus, if the condition number of is roughly , then we should expect around digits of accuracy in our computed solution.
The accuracy of the least squares problem is governed by its own condition number . We would hope that we can solve the least squares problem with an accuracy like the rule-of-thumb error bound (3) we had for linear systems of equations, namely a bound like . But this is not the kind of accuracy we get for the least squares problem when we solve it using the normal equations. Instead, we get accuracy like
(4)
By solving the normal equations we effectively square the condition number! Perhaps this is not surprising as the normal equations also more-or-less square the matrix by computing . This squared condition number drastically effects the accuracy of the computed solution. If the condition number of is , then the normal equations give us absolute nonsense for ; we expect to get no digits of the answer correct. Contrast this to above, where we were able to get correct digits in the solution to despite the condition number of being times larger than !
All of this would be just a sad fact of life for the least squares problem if the normal equations and their poor accuracy properties were the best we could do for the least squares problem. But we can do better! One can solve linear least squares problems by computing a so-called QR factorization of the matrix .5In MATLAB, the least squares problem can be solved with QR factorization by calling “A\b”. Without going into details, the upshot is that the least squares solution by a well-designed6One way of computing the QR factorization is by Gram–Schmidt orthogonalization, but the accuracy properties of this are poor too. A gold-standard way of computing the QR factorization by means of Householder reflectors, which has excellent accuracy properties. QR factorization requires a similar amount of time to solving the normal equations and has dramatically improved accuracy properties, achieving the desirable rule-of-thumb behavior7More precisely, the rule of thumb is like . So even if we solve the least squares problem with QR factorization, we still get a squared condition number in our error bound, but this condition number squared is multiplied by the residual , which is small if the least squares fit is good. The least squares solution is usually only interesting when the residual is small, thus justifying dropping it in the rule of thumb.
(5)
I have not described how the QR factorization is accurately computed nor how to use the QR factorization to solve least squares problems nor even what the QR factorization is. All of these topics are explained excellently by thestandardtextbooks in this area, as well as by publicly available resources like Wikipedia. There’s much more that can be said about the many benefits of solving the least squares problem with the QR factorization,8E.g., it can work for sparse matrices while the normal equations often do not, it has superior accuracy to Gaussian elimination with partial pivoting even for solving linear systems, the “” matrix in the QR factorization can be represented implicitly as a product of easy-to-compute-with Householder reflectors which is much more efficient when, etc. but in the interest of brevity let me just say this: TL;DR when presented in the wild with a least squares problem, the solution method one should default to is one based on a well-implemented QR factorization, not solving the normal equations.
Suppose for whatever reason we don’t have a high quality QR factorization algorithm at our disposal. Must we then resort to the normal equations? Even in this case, there is a way we can reduce the problem of solving a least squares problems to a linear system of equations without squaring the condition number! (For those interested, to do this, we recognize the normal equations as a Schur complement of a somewhat larger system of linear equations and then solve that. See Eq. (7) in this post for more discussion of this approach.)
The title of this post Don’t Solve the Normal Equations is deliberately overstated. There are times when solving the normal equations is appropriate. If is well-conditioned with a small condition number, squaring the condition number might not be that bad. If the matrix is too large to store in memory, one might want to solve the least squares problem using the normal equations and the conjugate gradient method.
However, the dramatically reduced accuracy of solving the normal equations should disqualify the approach from being the de-facto way of solving least squares problems. Unless you have good reason to think otherwise, when you see , solve a different way.
Suppose we have a thin, flat (two-dimensional) plate of homogeneous material and we measure the temperature at the border. What is the temperature inside the material? The solution to this problem is described by Laplace’s equation, one of the most ubiquitous partial differential equations in physics. Let denote the temperature of the material at point . Laplace’s equation states that, at any point on the interior of the material,
(1)
Laplace’s equation (1) and the specification of the temperature on the boundary form a well-posed mathematical problem in the sense that the temperature is uniquely determined at each point .1A well-posed problem is also required to depend continuously on the input data which, in this case, are the boundary temperatures. Indeed, the Laplace problem with boundary data is well-posed in this sense. We call this problem the Laplace Dirichlet problem since the boundary conditions
Another area of physics where the Laplace equation (1) appears is the study of electrostatics. In this case, represents the electric potential at the point . The Laplace Dirichlet problem is to find the electric potential in the interior of the region with knowledge of the potential on the boundary.
The electrostatic application motivates a different way of thinking about the Laplace equation. Consider the following question:
How would I place electric charges on the boundary to produce the electric potential at each point on the boundary?
This is a deliciously clever question. If I were able to find an arrangement of charges answering the question, then I could calculate the potential at each point in the interior by adding up the contribution to the electric potential of each element of charge on the boundary. Thus, I can reduce the problem of finding the electric potential at each point in the 2D region to finding a charge distribution on the 1D boundary to that region.
We shall actually use a slight variant of this charge distribution idea which differs in two ways:
Rather than placing simple charges on the boundary of the region, we place charge dipoles.2The reason for why this modification works better is an interesting question, but answering it properly would take us too far afield from the goals of this article.
Since we are considering a two-dimensional problem, we use a different formula for the electric potential than given by Coulomb’s law for charges in 3D. Also, since we are interested in solving the Laplace Dirichlet problem in general, we can choose a convenient dimensionless system of units. We say that the potential at a point induced by a unit “charge” at the origin is given by .
With these modifications, our new question is as follows:
How would I place a density of “charge” dipoles on the boundary to produce the electric potential at each point on the boundary?
We call this function the double layer potential for the Laplace Dirichlet problem. One can show the double layer potential satisfies a certain integral equation. To write down this integral equation, let’s introduce some more notation. Let be the region of interest and its boundary. Denote points concisely as vectors , with the length of denoted . The double layer potential satisfies
(2)
where the integral is taken over the surface of the region ; denotes the directional derivative taken in the direction normal (perpendicular) to the surface at the point . Note we choose a unit system for which hides physical constants particular to the electrostatic context, since we are interested in applying this methodology to the Laplace Dirichlet problem in general (possibly non-electrostatic) applications.
There’s one last ingredient: How do we compute the electric potential at points in the interior of the region? This is answered by the following formula:
(3)
The integral equation (2) is certainly nothing to sneeze at. Rather than trying to comprehend it in its full glory, we shall focus on a special case for the rest of our discussion. Suppose the region is a circular disk with radius centered at . The the partial derivative in the integrand in (2) then is readily computed for points and both on the boundary of the circle:
Substituting in (2) then gives
(4)
The Sherman–Morrison Formula
We are interested in solving the integral equation (4) to obtain an expression for the double-layer potential , as this will give us a solution formula for the Laplace Dirichlet problem. Ultimately, we accomplish this by using a clever trick. In an effort to make this trick seem more self-evident and less of a “rabbit out of a hat”, I want to draw an analogy to a seemingly unrelated problem: rank-one updates to linear systems of equations and the Sherman–Morrison formula.3In accordance with Stigler’s law of eponymy, the Sherman–Morrison formula was actually discovered by William J. Duncan five years before Sherman, Morrison, and Woodbury. For a more general perspective on the Sherman–Morrison formula and its generalization to the Sherman–Morrison–Woodbury formula, you may be interested in the following post of mine on Schur complements and block Gaussian elimination.
Suppose we want to solve the system of linear equations
(5)
where is an square matrix and , , and are length- vectors. We are ultimately interested in finding from . To gain insight into this problem, it will be helpful to first carefully considered the problem in reverse: computing from . We could, of course, perform this computation by forming the matrix in memory and multiplying it with , but there is a more economical way:
Form .
Compute .
Standing back, observe that we now have a system of equations for unknowns and . Specifically, our first equation can be rewritten as
which combined with the second equation
gives the by system4This “state space approach” of systematically writing out a matrix–vector multiply algorithm and then realizing this yields a larger system of linear equations was initially taught to be by my mentor Shiv Chandrasekaran; this approach has much more powerful uses, such as in the theory of rank-structured matrices.
(6)
The original equation for (5) can be derived from the “lifted” equation (6) by applying Gaussian elimination and eliminating the first row of the linear system (6). But now that we have the lifted equation (6), one can naturally wonder what would happen if we instead used Gaussian elimination to eliminate the last rows of (6); this will give us an equation for which we can solved without first computing . Doing this so-called block Gaussian elimination yields
This example shows how it can be conceptually useful to lift a linear system of equations by adding additional variables and equations and then “do Gaussian elimination in a different order”. The same insight shall be useful in solving integral equations like (4).
Solving for the Double Layer Potential
Let’s try repeating the playbook we executed for the rank-one-updated linear system (5) and apply it to the integral equation (4). We are ultimately interested in computing from but, as we did last section, let’s first consider the reverse. To compute from , we first evaluate the integral
Substituting this into (4) gives the system of equations
(7)
(8)
In order to obtain (4) from (7) and (8), we add times equation (8) to equation (7). Following last section, we now instead eliminate from equation (8) using equation (7). To do this, we need to integrate equation (7) in order to cancel the integral in equation (8):
Adding times this integrated equation to equation (8) yields
Thus plugging this expression for into equation (7) yields
We’ve solved for our double layer potential!
As promised, the double layer potential can be used to give a solution formula (known as the Poisson integral formula) for the Laplace Dirichet problem. The details are a mechanical, but also somewhat technical, exercise in vector calculus identities. We plug through the details in the following extra section.
Poisson Integral Formula
Let’s finish this up by using the double layer to derive a solution formula for the electric potential at a point in the interior of the region. To do this, we use equation (3):
(9)
We now need to do a quick calculation which is somewhat technical and not particularly enlightening. We evaluate using the divergence theorem:
Computing the boundary derivative for the spherical region centered at the origin with radius , we obtain the formula
We’ve succeeded at deriving a solution formula for for points in the interior of the disk in terms of for points on the boundary of the disk. This is known as the Poisson integral formula for the disk in two dimensions. This formula can be generalized to balls in higher dimensions, though this proof technique using “Sherman–Morrison” fails to work in more than two dimensions.
Sherman–Morrison for Integral Equations
Having achieved our main goal of deriving a solution formula for the 2D Laplace Dirichlet problem for a circular domain, I want to take a step back to present the approach from two sections ago in more generality. Consider a more general integral equation of the form
(10)
where is some region in space, , , and are functions of one or two arguments on , and is a nonzero constant. Such an integral equation is said to be of the second kind. The integral equation for the Laplace Dirichlet problem (2) is of this form with , , , and . We say the kernel is separable with rank if can be expressed in the form
With the circular domain, the Laplace Dirichlet integral equation (2) is separable with rank .5E.g., set and . We shall focus on the second kind integral equation (10) assuming the kernel is separable with rank (for simplicity, we set ):
(11)
Let’s try and write this equation in a way that’s more similar to the linear system of equation (5). To do this, we make use of linear operators defined on functions:
Let denote the identity operator on functions: It takes as inputs function and outputs the function unchanged.
Let denote the “integration against operator”: It takes as input a function and outputs the number .
With these notations, equation (11) can be written as
Using the same derivation which led to the Sherman–Morrison formula for linear systems of equations, we can apply the Sherman–Morrison formula to this integral equation in operator form, yielding
Therefore, the solution to the integral equation (11) is
This can be interpreted as a kind of Sherman–Morrison formula for the integral equation (11).
One can also generalize to provide a solution formula for the second-kind integral equation (10) for a separable kernel with rank ; in this case, the natural matrix analog is now the Sherman–Morrison–Woodbury identity rather than the Sherman–Morrison formula. Note that this solution formula requires the solution of a system of linear equations. One can use this as a numerical method to solve second-kind integral equations: First, we approximate by a separable kernel of a modest rank and then compute the exact solution of the resulting integral equation with the approximate kernel.6A natural question is why one might want to solve an integral equation formulation of a partial differential equations like the Laplace or Helmholtz equation. An answer is that that formulations based on second-kind integral equations tend to lead to systems of linear equations which much more well-conditioned as compared to other methods like the finite element method. They have a number of computational difficulties as well, as the resulting linear systems of equations are dense and may require elaborate quadrature rules to accurately compute.
My goal in writing this post was to discuss two topics which are both near and dear to my heart, integral equations and the Sherman–Morrison formula. I find the interplay of these two ideas to be highly suggestive. It illustrates the power of the analogy between infinite-dimensional linear equations, like differential and integral equations, and finite-dimensional ones, which are described by matrices. Infinite dimensions certainly do have their peculiarities and technical challenges, but it can be very useful to first pretend infinite-dimensional linear operators (like integral operators) are matrices, do calculations to derive some result, and then justify these computations rigorously post hoc.7The utility of this technique is somewhat of an open secret among some subset of mathematicians and scientists, but such heuristics are usually not communicated to students explicitly, at least in rigorous mathematics classes.
In this post, I want to discuss a beautiful and somewhat subtle matrix factorization known as the Vandermonde decomposition that appears frequently in signal processing and control theory. We’ll begin from the very basics, introducing the controls-and-signals context, how the Vandermonde decomposition comes about, and why it’s useful. By the end, I’ll briefly share how we can push the Vandermonde decomposition beyond matrices to the realm of tensors, which will can allow us to separate mixed signals from multiple measurements. This tensorial generalization plays an important role in my paper -decompositions, sparse component analysis, and the blind separation of sums of exponentials, joint work with Nithin Govindajaran and Lieven De Lathauwer, which recently appeared in the SIAM Journal of Matrix Analysis and Applications.
Finding the Frequencies
Suppose I give you a short recording of a musical chord consisting of three notes. How could you determine which three notes they were? Mathematically, we can represent such a three-note chord as a combination of scaled and shifted cosine functions
(1)
We are interested in obtaining the (angular) frequencies , , and .
In the extreme limit, when we are given the values of the signal for all , both positive and negative, the frequencies are immediately given by taking a Fourier transform of the function . In practice, we only have access to the function at certain times which we assume are equally spaced
Given the samples
we could try to identify , , and using a discrete Fourier transform.1The discrete Fourier transform can be computed very quickly using the fast Fourier transform, as I discussed in a previous post. Unfortunately, this generally requires a large number of samples to identify , , and accurately. (The accuracy scales roughly like , where is the number of samples.) We are interested in finding a better way to identify the frequencies.
Now that we’ve moved from the function , defined for any real input , to a set of samples it will be helpful to rewrite our formula (1) for in a different way. By Euler’s identity, the cosines can be rewritten as
As a consequence, we can rewrite one of the frequency components in (1) as
Here, and are complex coefficients and which contain the same information as the original parameters and . Now notice that we are only interest in values which are multiples of the spacing . Thus, our frequency component can be further rewritten as
where and . Performing these reductions, our samples take the form
(2)
We’ve now reformulated our frequency problems in identifying the parameters and in the relation (2) from a small number of measurements .
Frequency Finding as a Matrix Factorization
We will return to the algorithmic problem of identifying the parameters in the relation (2) from measurements in a little bit. First, we will see that (2) can actually be written as a matrix factorization. Understanding computations by matrix factorization has been an extremely successful paradigm in applied mathematics, and we will see in this post how viewing (2) as a matrix factorization can be very useful.
While it may seem odd at first,2As pointed out to me on math stack exchange, one reason forming the Hankel matrix is sensible is because it effectively augments the sequence of numbers into a sequence of vectors given by the columns of . This can reveal patterns in the sequence which are less obvious when it is represented as given just as numbers. For instance, any seven columns of are linearly dependent, a surprising fact since the columns of have length which can be much larger than seven. In addition, as we will soon effectively exploit later, vectors in the nullspace of (or related Hankel matrices derived from the sequence) give recurrence relations obeyed by the sequence. This speaks to a general phenomenon where properties of sequence (say, arising from snapshots of a dynamical system) can sometimes become more clear by this procedure of delay embedding. it will be illuminating to repackage the measurements as a matrix:
(3)
Here, we have assumed is odd. The matrix is known as the Hankel matrix associated with the sequence . Observe that the entry in position of depends only on the sum of the indices and , . (We use a zero-indexing system to label the rows and columns of where, for instance, the first row of is row .)
Let’s see how we can interpret the frequency decomposition (2) as a factorization of the Hankel matrix . We first write out using (2):
(4)
The power was just begging to be factorized as , which we did. Equation (4) almost looks like the formula for the product of two matrices with entries , so it makes sense to introduce the matrix with entry . This is a so-called Vandermonde matrix associated with and has the form
If we also introduce the diagonal matrix , the formula (4) for can be written as the matrix factorization3In the Vandermonde decomposition , the factor appears transposed even when is populated with complex numbers! This differs from the usual case in linear algebra where we use the conjugate transpose rather than the ordinary transpose when working with complex matrices. As a related issue, observe that if at least one of the measurements is a (non-real) complex number, the Hankel matrix is symmetric but not Hermitian.
(5)
This is the Vandermonde decomposition of the Hankel matrix , a factorization of as a product of the transpose of a Vandermonde matrix, a diagonal matrix, and that same Vandermonde matrix.
The Vandermonde decomposition immediately tells us all the information and describing our sampled recording via (2). Thus, the problem of determining and is equivalent to finding the Vandermonde decomposition (5) of the Hankel matrix .
Computing the Vandermonde Decomposition: Prony’s Method
Computing the Vandermonde decomposition accurately can be a surprisingly hard task, particularly if the measurements are corrupted by even a small amount of measurement error. In view of this, I want to present a very classical way of computing this decomposition (dating back to 1795!) known as Prony’s method. This method is conceptually simple and will be a vehicle to continue exploring frequency finding and its connection with Hankel matrices. It remainsinuse, though it’s accuracy may be significantly worse compared to other methods.
As a first step to deriving Prony’s method, let’s reformulate the frequency finding problem in a different way. Sums of cosines like the ones in our expression (1) for the function often appear as the solution to a (linear) ordinary differential equation (ODE). This means that one way we could find the frequencies comprising would be to find a differential equation which satisfies. Together with the initial condition , determining all the frequencies would be very straightforward.
Since we only have access to samples of at regular time intervals, we will instead look for the “discrete-time” analog of a linear ODE, a linear recurrence relation. This is an expression of the form
(6)
In our case, we’ll have because there are six terms in the formula (2) for . Together with initial conditions , such a recurrence will allow us to determine the parameters and in our formula (2) for our sampled recordings and hence also allow us to compute the Vandermonde decomposition (5).
Observe that the recurrence (6) is a linear equation in the variables . A very good rule of thumb in applied mathematics is to always write down linear equations in matrix–vector notation in see how it looks. Doing this, we obtain
(7)
Observe that the matrix on the right-hand side of this equation is also a Hankel matrix (like in (3)) formed from the samples . Call this Hankel matrix . Unlike in (3), is rectangular. If is much larger than , will be tall, possessing many more rows than columns. We assume going forward.4 would also be fine for our purposes, but we assume to illustrate this highly typical case.
Let’s write (7) a little more compactly as
(8)
where we’ve introduced for the vector on the left-hand side of (7) and collected the recurrence coefficients into a vector . For a typical system of linear equations like (8), we would predict the system to have no solution : Because has more rows than columns (if ), the system equations (8) has more equations than unknowns. Fortunately, we are not in the typical case. Despite the fact that we have more equations than unknowns, the linear equations (8) have a unique solution .5This solution can be computed by solving the system of linear equations In particular, the matrix on the right-hand side of this equation is guaranteed to be nonsingular under our assumptions. Using the Vandermonde decomposition, can you see why? The existence of a unique solution is a consequence of the fact that the samples satisfy the formula (2). As a fun exercise, you might want to verify the existence of a unique satisfying (8)!
As a quick aside, if the measurements are corrupted by small measurement errors, then the equations (8) will usually not possess a solution. In this case, it would be appropriate to find the least squares solution to equation (8) as a way of mitigating these errors.
Hurrah! We’ve found the coefficients providing a recurrence relation (6) for our measurements . All that is left is to find the parameters and in our signal formula (2) and the Vandermonde decomposition (5). Fortunately, this is just a standard computation for linear recurrence relations, nicely paralleling the solution of (homogenous) linear ODEs by means of the so-called “characteristic equation”. I’ll go through fairly quickly since this material is well-explained elsewhere on the internet (like Wikipedia). Let’s guess that our recurrence (6) has a solution of the form ; we seek to find all complex numbers for which this is a bonafide solution. Plugging this solution into the formula (6) for gives
(9)
This is the so-called characteristic equation for the recurrence (6). As a single-variable polynomial equation of degree six, it has six complex solutions . These numbers are precisely those numbers which appear in the sequence formula (2) and the Vandermonde decomposition (5).
Finally, we need to compute the coefficients . But this is easy. Observe that the formula (2) provides the following system of linear equations for :
(10)
Again, this system of equations will have a unique solution if the measurements are uncorrupted by errors (and can be solved in the least squares sense if corrupted). This gives , completing our goal of computing the parameters in the formula (2) or, equivalently, finding the Vandermonde decomposition (5).
We have accomplished our goal of computing the Vandermonde decomposition. The approach by which we did so is known as Prony’s method, as mentioned in the introduction to this section. As suggested, this method may not always give high-accuracy results. There are two obvious culprits that jump out about why this is the case. Prony’s method requires solving for the roots of the polynomial equation (9) expressed “in the monomial basis” and solving a system of linear equations (10) with a (transposed) Vandermonde matrix. Both of theseproblems can be notoriously ill-conditioned and thus challenging to solve accurately and may require the measurements to be done to very high accuracy. Notwithstanding this, Prony’s method does useful results in some cases and forms the basis for potentially more accurate methods, such as those involving generalized eigenvalue problems.
Separating Signals: Extending the Vandermonde Decomposition to Tensors
In our discussion of the frequency identification problem, the Vandermonde decomposition (5) has effectively been an equivalent way of showing the samples are a combination of exponentials . So far, the benefits of the matrix factorization perspective have yet to really reveal themselves.
So what are the benefits of the Vandermonde decompostions? A couple of nice observations related to the Vandermonde decomposition and the “Hankelization” of the signals have already been lurking in the background. For instance, the rank of the Hankel matrix is the number of frequency components needed to describe the samples and the representation of the samples as a mixture of exponentials is uniquely determined only if the matrix does not have full rank; I have a little more to say about this at the very end. There are also benefits to certain computational problems; one can use Vandermonde decompositions to compute super high accuracy singular value decompositions of Hankel matrices.
The power of the Vandermonde decomposition really starts to shine when we go beyond the basic frequency finding problem we discussed by introducing more signals. Suppose now there are three short recordings , , and . (Here, the superscript denotes an index rather than differentiation.) Each signal is a weighted mixture of three sources, , and , each of which plays a musical chord of three notes (thus representable as a sum of cosines as in (1)). One can think of the sources of being produced three different musical instruments at different places in a room and the recordings , , and being taken from different microphones in the room.6This scenario of instruments and microphones ignores the finite propagation speed of sound, which also would introduce time delays in the sources in the recorded signals. We effectively treat the speed of sound as being instantaneous. Our goal is now not just to identify the musical notes in the recordings but also to identify how to assign those notes to reconstruct the source signals , , and .
Taking inspiration from earlier, we record samples for each recording and form each collection of samples into a Hankel matrix
Here comes the crazy part: Stack the Hankelized recordings , , and as slices of a tensor. A tensor, in this context, just means a multidimensional array of numbers. Just as a vector is a one-dimensional array and a matrix is a two-dimensional array, a tensor could have any number of dimensions. In our case, we need just three. If we use a MATLAB-esque indexing notation, is a array given by
The remarkable thing is that the source signals can be determined (under appropriate conditions) by computing a special kind of Vandermonde decomposition of the tensor! (Specifically, the required decomposition is a Vandermonde-structured -block term decomposition of the tensor .) Even more cool, this decomposition can be computed using general-purpose software like Tensorlab.
If this sounds interesting, I would encourage you to check out my recently published paper -decompositions, sparse component analysis, and the blind separation of sums of exponentials, joint work with Nithin Govindajaran and Lieven De Lathauwer and recently published in the SIAM Journal on Matrix Analysis and Applications. In the paper, we explain what this -decomposition is and how applying it to can be used to separate mixtures of exponentials signals from the resulting Vandermonde structure, an idea originating in the work of De Lathauewer. A very important question for these signal separation problems is that of uniqueness. Given the three sampled recordings (comprising the tensor ), is there just one way of unscrambling the mixtures into different sources or multiple? If there are multiple, then we might have possibly computed the wrong one. If there is just a single unscrambling, though, then we’ve done our job and unmixed the scrambled signals. The uniqueness of these tensor decompositions is fairly complicated math, and we survey existing results and prove new ones in this paper.7One of our main technical contributions is a new notion of uniqueness of -decompositions which we believe is nicely adapted to the signal separation context. Specfically, we prove mathematized versions of the statement “if the source signals are sufficiently different from each others and the measurements of sufficiently high quality, then the signals can uniquely be separated”.
Conclusions, Loose Ends, and Extensions
The central idea that we’ve been discussing is how it can be useful to convert between a sequence of observations and a special matricization of this sequence into a Hankel matrix (either square, as in (3), or rectangular, as in (7)). By manipulating the Hankel matrix, say, by computing its Vandermonde decomposition (5), we learn something about the original signal, namely a representation of the form (2).
This is a powerful idea which appears implicitly or explicitly throughout various subfields of mathematics, engineering, and computation. As with many other useful ideas, this paradigm admits many natural generalizations and extensions. We saw one already in this post, where we extended the Vandermonde decomposition to the realm of tensors to solve signal separation problems. To end this post, I’ll place a few breadcrumbs at the beginning of a few of the trails of these generalizations for any curious to learn more, wrapping up a few loose ends on the way.
Is the Vandermonde Decomposition Unique?
A natural question is whether the Vandermonde decomposition (5) is unique. That is, is it possible that there exists two Vandermonde decompositions
of the same (square) Hankel matrix ? This is equivalent to whether the frequency components can be uniquely determined from the measurements .
Fortunately, the Vandermonde decomposition is unique if (and only if) the matrix is a rank-deficient matrix. Let’s unpack this a little bit. (For those who could use a refresher on rank, I have a blog post on precisely this topic.) Note that the Vandermonde decomposition is a rank factorization8Rank factorizations are sometimes referred to as “rank-revealing factorizations”. I discuss my dispreference for this term in my blog post on low-rank matrices. since has rows, has full (row) rank, and is invertible. This means that if take enough samples of a function which is a (finite) combinations of exponentials, the matrix will be rank-deficient and the Vandermonde decomposition unique.9The uniqueness of the Vandermonde decomposition can be proven by showing that, in our construction by Prony’s method, the ‘s, ‘s, and ‘s are all uniquely determined. If too few samples are taken, then does not contain enough information to determine the frequency components of the signal and thus the Vandermonde decomposition is non-unique.
Does Every Hankel Matrix Have a Vandermonde Decomposition?
This post has exclusively focused on a situation where we are provided with sequence we know to be represented as a mixture of exponentials (i.e., taking the form (2)) from which the existence of the Vandermonde decomposition (5) follows immediately. What if we didn’t know this were the case, and we were just given a (square) Hankel matrix . Is guaranteed to possess a Vandermonde decomposition of the form (5)?
Unfortunately, the answer is no; there exist Hankel matrices which do not possess a Vandermonde decomposition. The issue is related to the fact that the appropriate characteristic equation (analogous to (9)) might possess repeated roots, making the solutions to the recurrence (6) not just take the form but also and perhaps , , etc.
Are There Cases When the Vandermonde Decomposition is Guaranteed To Exist?
There is one natural case when a (square) Hankel matrix is guaranteed to possess a Vandermonde decomposition, namely when the matrix is nonsingular/invertible/full-rank. Despite this being a widely circulated fact, I am unaware of a simple proof for why this is the case. Unfortunately, there is not just one but infinitely many Vandermonde decompositions for a nonsingular Hankel matrix, suggesting these decompositions are not useful for the frequency finding problem that motivated this post.
What If My Hankel Matrix Does Not Possess a Vandermonde Decomposition?
As discussed above, a Hankel matrix may fail to have a Vandermonde decomposition if the characteristic equation (a la (9)) has repeated roots. This is very much analogous to the case of a non-diagonalizable matrix for which the characteristic polynomial has repeated roots. In this case, while diagonalization is not possible, one can “almost-diagonalize” the matrix by reducing it to its Jordan normal form. In total analogy, every Hankel matrix can be “almost Vandermonde decomposed” into a confluent Vandermonde decomposition (a discovery that appears to have been made independentlyseveraltimes). I will leave these links to discuss the exact nature of this decomposition, though I warn any potential reader that these resources introduce the decomposition first for Hankel matrices with infinitely many rows and columns before considering the finite case as we have. One is warned that while the Vandermonde decomposition is always a rank decomposition, the confluent Vandermonde decomposition is not guaranteed to be one.10Rather, the confluent Vandermonde decomposition is a rank decomposition for an infinite extension of a finite Hankel matrix. Consider the Hankel matrix This matrix has rank-two but no rank-two confluent Vandermonde decomposition. The issue is that when extended to an infinite Hankel matrix this (infinite!) matrix has a rank exceeding the size of the original Hankel matrix .
The Toeplitz Vandermonde Decomposition
Just as it proved useful to arrange samples into a Hankel matrix, it can also be useful to form them into a Toeplitz matrix
One can interconvert between Hankel and Toeplitz matrices by reversing the order of the rows. As such, to the extent to which Hankel matrices possess Vandermonde decompositions (with all the asterisks and fine print just discussed), Toeplitz matrices do as well but with the rows of the first factor reversed:
There is a special and important case where more is true. If a Toeplitz matrix is (Hermitian) positive semidefinite, then always possesses a Vandermonde decomposition of the form
where is a Vandermonde matrix associated with parameters which are complex numbers of absolute value one and is a diagonal matrix with real positive entries.12The keen-eyed reader will note that appears conjugate transposed in this formula rather than transposed as in the Hankel Vandermonde decomposition (5). This Vandermonde decomposition is unique if and only if is rank-deficient. Positive semidefinite Toeplitz matrices are important as they occur as autocorrelation matrices which effectively describe the similarity between a signal and different shifts of itself in time. Autocorrelation matrices appear under different names in everything from signal processing to random processes to near-term quantum algorithms (a topic near and dear to my heart). A delightfully simple and linear algebraic derivation of this result is given by Yang and Xie (see Theorem 1).13Unfortunately, Yang and Xie incorrectly claim that every Toeplitz matrix possesses a rank factorization Vandermonde decomposition of the form where is a Vandermonde matrix populated with entries on the unit circle and is a diagonal matrix of possibly *complex* entries. This claim is disproven by the example . This decomposition can be generalized to infinite positive semidefinite Toeplitz matrices (appropriately defined).14Specifically, one can show that an infinite positive semidefinite Toeplitz matrix (appropriately defined) also has a “Vandermonde decomposition” (appropriately defined). This result is often known as Herglotz’s theorem and is generalized by the Bochner–Weil theorem.
In this post, I want to discuss a beautiful and simple geometric picture of the perturbation theory of definite generalized eigenvalue problems. As a culmination, we’ll see a taste of the beautiful perturbation theory of Mathias and Li, which appears to be not widely known in virtue of only being explained in a technical report. Perturbation theory for the generalized eigenvalue problem is a bit of a niche subject, but I hope you will stick around for some elegant arguments. In addition to explaining the mathematics, I hope this post serves as an allegory for the importance of having the right way of thinking about a problem; often, the solution to a seemingly unsolvable problem becomes almost self-evident when one has the right perspective.
What is a Generalized Eigenvalue Problem?
This post is about the definite generalized eigenvalue problem, so it’s probably worth spending a few words talking about what generalized eigenvalue problems are and why you would want to solve them. Slightly simplifying some technicalities, a generalized eigenvalue problem consists of finding nonzero vectors and a (possibly complex) numbers such that .1In an unfortunate choice of naming, there is actually a completely different sense in which it makes sense to talk about generalized eigenvectors, in the context of the Jordan normal form for standard eigenvalue problems. The vector is called an eigenvector and its eigenvalue. For our purposes, and will be real symmetric (or even complex Hermitian) matrices; one can also consider generalized eigenvalue problemss for nonsymmetric and even non-square matrices and , but the symmetric case covers many applications of practical interest. The generalized eigenvalue problem is so-named because it generalizes the standard eigenvalue problem , which is a special case of the generalized eigenvalue problem with .2One can also further generalize the generalized eigenvalue problem to polynomial and nonlinear eigenvalue problems.
Why might we want so solve a generalized eigenvalue problem? The applications are numerous (e.g., in chemistry, quantum computation, systems and control theory, etc.). My interest in perturbation theory for generalized eigenvalue problems arose in the analysis of a quantum algorithm for eigenvalue problems in chemistry, and the theory discussed in this article played a big role in that analysis. To give an application which is more easy to communicate than the quantum computation application which motivated my own interest, let’s discuss an application in classical mechanics.
The Lagrangian formalism is a way of reformulating Newton’s laws of motion in a general coordinate system.3The benefits of the Lagrangian framework are far deeper than working in generalized coordinate systems, but this is beyond the scope of our discussion and mostly beyond the scope of what I am knowledgeable enough to write meaningfully about. If denotes a vector of generalized coordinates describing our system and denotes ‘s time derivative, then the time evolution of a system with Lagrangian functional are given by the Euler–Lagrange equations. If we choose to represent the deviation of our system from equilibrium,4That is, in our generalized coordinate system, is a (static) equilibrium configuration for which whenever and . then our Lagrangian is well-approximated by it’s second order Taylor series:
By the Euler–Lagrange equations, the equations of motion for small deviations from this equillibrium point are described by
A fundamental set of solutions of this system of differential equations is given by , where and are the generalized eigenvalues and eigenvectors of the pair .5That is, all solutions to can be written (uniquely) as linear combinations of solutions of the form . In particular, if all the generalized eigenvalues are positive, then the equillibrium is stable and the square roots of the eigenvalues represent the modes of vibration. In the most simple mechanical systems, such as masses in one dimension connected by springs with the natural coordinate system, the matrix is diagonal with diagonal entries equal to the different masses. In even slightly more complicated “freshman physics” problems, it is quite easy to find examples where, in the natural coordinate system, the matrix is nondiagonal.6Almost always, the matrix is positive definite. As this example shows, generalized eigenvalue problems aren’t crazy weird things since they emerge as natural descriptions of simple mechanical systems like coupled pendulums.
One reason generalized eigenvalue problems aren’t more well-known is that one can easily reduce a generalized eigenvalue problem into a standard one. If the matrix is invertible, then the generalized eigenvalues of are just the eigenvalues of the matrix . For several reasons, this is a less-than-appealing way of reducing a generalized eigenvalue problem to a standard eigenvalue problem. A better way, appropriate when and are both symmetric and is positive definite, is to reduce the generalized eigenvalue problem for to the symmetrically reduced matrix , which also possesses the same eigenvalues as . In particular, the matrix remains symmetric, which shows that has real eigenvalues by the spectral theorem. In the mechanical context, one can think of this reformulation as a change of coordinate system in which the “mass matrix” becomes the identity matrix .
There are several good reasons to not simply reduce a generalized eigenvalue problem to a standard one, and perturbation theory gives a particular good reason. In order for us to change coordinates to change the matrix into an identity matrix, we must first know the matrix. If I presented you with an elaborate mechanical system which you wanted to study, you would need to perform measurements to determine the and matrices. But all measurements are imperfect and the entries of and are inevitably corrupted by measurement errors. In the presence of these measurement errors, we must give up on computing the normal modes of vibration perfectly; we must content ourselves with computing the normal modes of vibration plus-or-minus some error term we hope we can be assured is small if our measurement errors are small. In this setting, reducing the problem to seems less appealing, as I have to understand how the measurement errors in and are amplified in computing the triple product . This also suggests that computing may be a poor algorithmic strategy in practice: if the matrix is ill-conditioned, there might be a great deal of error amplification in the computed product . One might hope that one might be able to devise algorithms with better numerical stability properties if we don’t reduce the matrix pair to a single matrix. This is not to say that reducing a generalized eigenvalue problem to a standard one isn’t a useful tool—it definitely is. However, it is not something one should do reflexively. Sometimes, a generalized eigenvalue problem is best left as is and analyzed in its native form.
The rest of this post will focus on the question if and are real symmetric matrices (satisfying a definiteness condition, to be elaborated upon below), how do the eigenvalues of the pair compare to those of , where and are small real symmetric perturbations? In fact, it shall be no additional toil to handle the complex Hermitian case as well while we’re at it, so we shall do so. (Recall that a Hermitian matrix satisfies , where is the conjugate transpose. Since the complex conjugate does not change a real number, a real Hermitian matrix is necesarily symmetric .) For the remainder of this post, let , , , and be Hermitian matrices of the same size. Let and denote the perturbations.
Symmetric Treatment
As I mentioned at the top of this post, our mission will really be to find the right way of thinking about perturbation theory for the generalized eigenvalue problem, after which the theory will follow much more directly than if we were to take a frontal assault on the problem. As we go, we shall collect nuggets of insight, each of which I hope will follow quite naturally from the last. When we find such an insight, we shall display it on its own line.
The first insight is that we can think of the pair and interchangeably. If is a nonzero eigenvalue of the pair , satisfying , then . That is, is an eigenvalue of the pair . Lack of symmetry is an ugly feature in a mathematical theory, so we seek to smooth it out. After thinking a little bit, notice that we can phrase the generalized eigenvalue condition symmetrically as with the associated eigenvalue being given by . This observation may seem trivial at first, but let us collect it for good measure.
Treat and symmetrically by writing the eigenvalue as with .
Before proceeding, let’s ask a question that, in our new framing, becomes quite natural: what happens when ? The case is problematic because it leads to a division by zero in the expression . However, if we have , this expression still makes sense: we’ve found a vector for which but . It makes sense to consider still an eigenvector of with eigenvalue ! Dividing by zero should justifiably make one squeemish, but it really is quite natural in the case to treat as a genuine eigenvector with eigenvalue .
Things get even worse if we find a vector for which . Then, any can reasonably considered an eigenvalue of since . In such a case, all complex numbers are simultaneously eigenvalues of , in which case we call singular.7More precisely, a pair is singular if the determinant is identically zero for all . For the generalized eigenvalue problem to make sense for a pair , it is natural to require that not be singular. In fact, we shall assume an even stronger “definiteness” condition which ensures that has only real (or infinite) eigenvalues. Let us return to this question of definiteness in a moment and for now assume that is not singular and possesses real eigenvalues.
With this small aside taken care of, let us return to the main thread. By modeling eigenvalues as pairs , we’ve traded one ugliness for another. While reformulating the eigenvalue as a pair treats and symmetrically, it also adds an additional indeterminacy, scale. For instance, if is an eigenvalue of , then so is . Thus, it’s better not to think of so much as a pair of numbers together with all of its possible scalings.8Projective space provides a natural framework for studying such vectors up to scale indeterminacy. For reasons that shall hopefully become more clear as we go forward, it will be helpful to only consider all the possible positive scalings of —e.g., all for . Geometrically, the set of all positive scalings of a point in two-dimensional space is precisely just a ray emanating from the origin.
Represent eigenvalue pairs as rays emanating from the origin to account for scale ambiguity.
Now comes a standard eigenvalue trick. It’s something you would never think to do originally, but once you see it once or twice you learn to try it as a matter of habit. The trick: multiply the eigenvalue-eigenvector relation by the (conjugate) transpose of :9For another example of the trick, try applying it to the standard eigenvalue problem . Multiplying by and rearranging gives —the eigenvalue is equal to the expression , which is so important it is given a name: the Rayleigh quotient. In fact, the largest and smallest eigenvalues of can be found by maximizing and minimizing the Rayleigh quotient.
The above equation is highly suggestive: since and are only determined up to a scaling factor, it shows we can take and . And by different scalings of the eigenvector , we can scale and by any positive factor we want. (This retroactively shows why it makes sense to only consider positive scalings of and .10To make this point more carefully, we shall make great use of the identification between pairs and the pair of quadratic forms . Thus, even though and lead to equivalent eigenvalues since , and don’t necessarily both arise from a pair of quadratic forms: if , this does not mean there exists such that . Therefore, we only consider equivalent to if .) The expression is so important that we give it a name: the quadratic form (associated with and evaluated at ).
The eigenvalue pair can be taken equal to the pair of quadratic forms .
Complexifying Things
Now comes another standard mathematical trick: represent points in two-dimensional space by complex numbers. In particular, we identify the pair with the complex number .11Recall that we are assuming that is real, so we can pick a scaling in which both and are real numbers. Assume we have done this. Similar to the previous trick, it’s not fully clear why this will pay off, but let’s note it as an insight.
Identify the pair with the complex number .
Now, we combine all the previous observations. The eigenvalue is best thought of as a pair which, up to scale, can be taken to be and . But then we represent as the complex number
Let’s stop for a moment and appreciate how far we’ve come. The generalized eigenvalue problem is associated with the expression .If we just went straight from one to the other, this reduction would appear like some crazy stroke of inspiration: why would I ever think to write down ? However, just following our nose lead by a desire to treat and symmetrically and applying a couple standard tricks, this expression appears naturally. The expression will be very useful to us because it is linear in and , and thus for the perturbed problem , we have that : consequently, is a small perturbation of . This observation will be very useful to us.
If is the eigenvector, then the complex number is .
Definiteness and the Crawford Number
With these insights in hand, we can now return to the point we left earlier about what it means for a generalized eigenvalue problem to be “definite”. We know that if there exists a vector for which , then the problem is singular. If we multiply by , we see that this means that as well and thus . It is thus quite natural to assume the following definiteness condition:
The pair is said to be definite if for all complex nonzero vectors .
A definite problem is guaranteed to be not singular, but the reverse is not necessarily true; one can easily find pairs which are not definite and also not singular.12For example, consider . is not definite since for . However, is not singular; the only eigenvalue of the pair is and is not identically zero.. (Note does not imply unless and are both positive (or negative) semidefinite.)
The “natural” symmetric condition for to be “definite” is for for all vectors .
Since the expression is just scaled by a positive factor by scaling the vector , it is sufficient to check the definiteness condition for only complex unit vectors . This leads naturally to a quantitative characterization of the degree of definiteness of a pair :
The Crawford number13The name Crawford number was coined by G. W. Stewart in 1979 in recognition of Crawford’s pioneering work on the perturbation theory of the definite generalized eigenvalue problem. of a pair is the minimum value of over all complex unit vectors .
The Crawford number naturally quantifies the degree of definiteness.14In fact, it has been shown that the Crawford number is, in a sense, the distance from a definite matrix pair to a pair which is not simultaneously diagonalizable by congruence. A problem which has a large Crawford number (relative to a perturbation) will remain definite after perturbation, whereas the pair may become indefinite if the size of the perturbation exceeds the Crawford number. Geometrically, the Crawford number has the following interpretation: must lie on or outside the circle of radius centered at for all (complex) unit vectors .
The “degree of definiteness” can be quantified by the Crawford number .
Now comes another step in our journey which is a bit more challenging. For a matrix (in our case ), the set of complex numbers for all unit vectors has been the subject of considerable study. In fact, this set has a name
The field of values of a matrix is the set .
In particular, the Crawford number is just the absolute value of the closest complex number in the field of values to zero.
It is a very cool and highly nontrivial fact (called the Toeplitz–Hausdorff Theorem) that the field of values is always a convex set, with every two points in the field of values containing the line segment connecting them. Thus, as a consequence, the field of values for a definite matrix pair is always on “one side” of the complex plane (in the sense that there exists a line through zero which lies strictly on one side of15This is a consequence of the hyperplane separation theorem together with the fact that .).
The numbers for unit vectors lie on one half of the complex plane.
The field of values lies outside the circle of radius centered at and thus on one side of the complex plane.
From Eigenvalues to Eigenangles
All of this geometry is lovely, but we need some way of relating it to the eigenvalues. As we observed a while ago, each eigenvalue is best thought of as a ray emanating from the origin, owing to the fact that the pair can be scaled by an arbitrary positive factor. A ray is naturally associated with an angle, so it is natural to characterize an eigenvalue pair by the angle describing its arc.
But the angle of a ray is only defined up additions by full rotations ( radians). As such, to associate each ray a unique angle we need to pin down this indeterminacy in some fixed way. Moreover, this indeterminacy should play nice with the field of values and the field of values of the perturbation. But in the last section, we say that each of these field of angles lies (strictly) on one half of the complex plane. Thus, we can find a ray which does not intersect either field of values!
One possible choice is to measure the angle from this ray. We shall make a slightly different choice which plays better when we treat as a complex number . Recall that a number is an argument for if for some real number . The argument is multi-valued since is an argument for as long as is (for all integers ). However, once we exclude our ray , we can assign each complex number not on this ray a unique argument which depends continuously on . Denote this “branch” of the argument by . If represents an eigenvalue , we call an eigenangle.
Represent an eigenvalue pair by its associated eigenangle .
How are these eigenangles related to the eigenvalues? It’s now a trigonometry problem:
The eigenvalues are the cotangents of the eigenangles!
The eigenvalue is the cotangent of the eigenangle .
Variational Characterization
Now comes another difficulty spike in our line of reasoning, perhaps the largest in our whole deduction. To properly motivate things, let us first review some facts about the standard Hermitian/symmetric eigenvalue problem. The big idea is that eigenvalues can be thought of as the solution to a certain optimization problem. The largest eigenvalue of a Hermitian/symmetric matrix is given by the maximization problem
The largest eigenvalue is the maximum of the quadratic form over unit vectors . What about the other eigenvalues? The answer is not obvious, but the famous Courant–Fischer Theorem shows that the th largest eigenvalue can be written as the following minimax optimization problem
The minimum is taken over all subspaces of dimension whereas the maximum is taken over all unit vectors within the subspace . Symmetrically, one can also formulate the eigenvalues as a max-min optimization problem
These variational/minimax characterizations of the eigenvalues of a Hermitian/symmetric matrix are essential to perturbation theory for Hermitian/symmetric eigenvalue problems, so it is only natural to go looking for a variational characterization of the generalized eigenvalue problem. There is one natural way of doing this that works for positive definite: specifically, one can show that
This characterization, while useful in its own right, is tricky to deal with because it is nonlinear in and . It also treats and non-symmetrically, which should set off our alarm bells that there might be a better way. Indeed, the ingenious idea, due to G. W. Stewart in 1979, is to instead provide a variational characterization of the eigenangles! Specifically, Stewart was able to show16Stewart’s original definition of the eigenangles differs from ours; we adopt the definition of Mathias and Li. The result amounts to the same thing.
(1)
for the eigenangles .17Note that since the cotangent is decreasing on , this means that the eigenvalues are now in increasing order, in contrast to our convention from earlier in this section. This shows, in particular, that the field of values is subtended by the smallest and largest eigenangles.
The eigenangles satisfy a minimax variational characterization.
How Big is the Perturbation?
We’re tantalizingly close to our objective. The final piece in our jigsaw puzzle before we’re able to start proving perturbation theorems is to quantify the size of the perturbing matrices and . Based on what we’ve done so far, we see that the eigenvalues are natural associated with the complex number , so it is natural to characterize the size of the perturbing pair by the distance between and . But the difference between these two quantities is just
We’re naturally led to the question: how big can be? If the vector has a large norm, then quite large, so let’s fix to be a unit vector. With this assumption in place, the maximum size of is simple the distance of the farthest point in the field of values from zero. This quantity has a name:
The numerical radius of a matrix (in our case ) is .18This maximum is taken over all complex unit vectors .
The size of the perturbation is the numerical radius .
It is easy to upper-bound the numerical radius by more familiar quantities. For instance, once can straightforwardly show the bound , where is the spectral norm. We prefer to state results using the numerical radius because of its elegance: it is, in some sense, the “correct” measure of the size of the pair in the context of this theory.
Stewart’s Perturbation Theory
Now, after many words of prelude, we finally get to our first perturbation theorem. With the work we’ve done in place, the result is practically effortless.
Let denote the eigenangles of the perturbed pair and consider the th eigenangle. Let be the subspace of dimension achieving the minimum in the first equation of the variational principle (1) for the original unperturbed pair. Then we have
(2)
This is something of a standard trick when dealing with variational problems in matrix analysis: take the solution (in this case the minimizing subspace) for the original problem and plug it in for the perturbed problem. The solution may no longer be optimal, but it at least gives an upper (or lower) bound. The complex number must lie at least a distance from zero and . We’re truly toast if the perturbation is large enough to perturb to be equal to zero, so we should assume that .
For our perturbation theory to work, we must assume .
lies on or outside the circle centered at zero with radius . might lie anywhere in a circle centered at with radius , so one must have to ensure the perturbed problem is nonsingular (equivalently for every ).
Making the assumption that , bounding the right-hand side of (2) requires finding the most-counterclockwise angle necessary to subtend a circle of radius centered at , which must lie a distance from the origin. The worst-case scenario is when is exactly a distance from the origin, as is shown in the following diagram.
In the worst case, lies on the circle centered at zero with radius , which is subtended above by angle .
Solving the geometry problem for the counterclockwise-most subtending angle in this worst-case sitation, we conclude the eigenangle bound . An entirely analogous argument using the max-min variational principle (1) proves an identical lower bound, thus showing
(3)
In the language of eigenvalues, we have19I’m being a little sloppy here. For a result like this to truly hold, I believe all of the perturbed and unperturbed eigenangles should all be contained in one half of the complex plane.
Interpreting Stewart’s Theory
After much work, we have finally proven our first generalized eigenvalue perturbation theorem. After taking a moment to celebrate, let’s ask ourselves: what does this result tell us?
Let’s start with the good. This result shows us that if the perturbation, measured by the numerical radius , is much smaller than the definiteness of the original problem, measured by the Crawford number , then the eigenangles change by a small amount. What does this mean in terms of the eigenvalues? For small eigenvalues (say, less than one in magnitude), small changes in the eigenangles also lead to small changes of the eigenvalues. However, for large eigenangles, small changes in the eigenangle are magnified into potentially large changes in the eigenvalues. One can view this result in a positive or negative framing. On the one hand, large eigenvalues could be subject to dramatic changes by small perturbations; on the other hand, the small eigenvalues aren’t “taken down with the ship” and are much more well-behaved.
Stewart’s theory is beautiful. The variational characterization of the eigenangles (1) is a master stroke and exactly the extension one would want from the standard Hermitian/symmetric theory. From the variational characterization, the perturbation theorem follows almost effortlessly from a little trigonometry. However, Stewart’s theory has one important deficit: the Crawford number. All that Stewart’s theory tells is that all of the eigenangles change by at most roughly “perturbation size over Crawford number”. If the Crawford number is quite small since the problem is nearly indefinite, this becomes a tough pill to swallow.
The Crawford number is in some ways essential: if the perturbation size exceeds the Crawford number, the problem can become indefinite or even singular. Thus, we have no hope of fully removing the Crawford number from our analysis. But might it be the case that some eigenangles change by much less than “pertrubation size over Crawford number”? Could we possibly improve to a result of the form “the eigenangles change by roughly perturbation size over something (potentially) much less than the Crawford number”? Sun improved Stewart’s analysis in 1982, but the scourge of the Crawford number remained.20Sun’s bound does not explicitly have the Crawford number, instead using the quantity and another hard-to-concisely describe quantity. In many cases, one has nothing better to do than to bound , in which case the Crawford number has appeared again. The theory of Mathias and Li, published in a technical report in 2004, finally produced a bound where the Crawford number is replaced.
The Mathias–Li Insight and Reduction to Diagonal Form
Let’s go back to the Stewart theory and look for a possible improvement. Recall in the Stewart theory that we considered the point on the complex plane. We then argued that, in the worst case, this point would lie a distance from the origin and then drew a circle around it with radius . To improve on Stewart’s bound, we must somehow do something better than using the fact that . The insight of the Mathias–Li theory is, in some sense, as simple as this: rather than using the fact that (as in Stewart’s analysis), use how far actually is from zero, where is chosen to be the unit norm eigenvectors of .21This insight is made more nontrivial by the fact that, in the context of generalized eigenvalue problems, it is often not convenient to choose the eigenvectors to have unit norm. As Mathias and Li note, there are often two more popular normalizations for . If is positive definite, one often normalizes such that —the eigenvectors are thus made “-orthonormal”, generalizing the fact that the eigenvectors of a Hermitian/symmetric matrix are orthonormal. Another popular normalization is to scale such that . In this way, just taking the eigenvector to have unit norm is already a nontrivial insight.
Before going further, let us quickly make a small reduction which will simplify our lives greatly. Letting denote a matrix whose columns are the unit-norm eigenvectors of , one can verify that and are diagonal matrices with entries and respectively. With this in mind, it can make our lives a lot easy to just do a change of variables and (which in turn sends and ). The change of variables is very common in linear algebra and is called a congruence transformation.
Perform a change of variables by a congruence transformation with the matrix of eigenvectors.
While this change of variables makes our lives a lot easier, we must first worry about how this change of variables might effect the size of the perturbation matrices . It turns out this change of variables is not totally benign, but it is not maximally harmful either. Specifically, the spectral radius can grow by as much as a factor of .22This is because, in virtue of having unit-norm columns, the spectral norm of the matrix is . Further, note the following variational characterization of the spectral radius . Plugging these two facts together yields . This factor of isn’t great, but it is much better than if the bound were to degrade by a factor of the condition number , which can be arbitrarily large.
This change of variables may increase by at most a factor of .
From now on, we shall tacitly assume that this change of variables has taken place, with and being diagonal and and being such that is at most a factor larger than it was previously. We denote by and the th diagonal element of and , which are given by and where is the th unit-norm eigenvector
Mathias and Li’s Perturbation Theory
We first assume the perturbation is smaller than the Crawford number in the sense , which is required to be assured that the perturbed problem does not lose definiteness. This will be the only place in this analysis where we use the Crawford number.
Draw a circle of radius around .
If is the associated eigenangle, then this circle is subtended by arcs with angles
It would be nice if the perturbed eigenangles were guaranteed to lie in these arcs (i.e., ). Unfortunately this is not necessarily the case. If one is close to the origin, it will have a large arc which may intersect with other arcs; if this happens, we can’t guarantee that each perturbed eigenangle will remain within its individual arc. We can still say something though.
What follows is somewhat technical, so let’s start with the takeaway conclusion: is larger than any of the lower bounds . In particular, this means that is larger than the th largest of all the lower bounds. That is, if we rearrange the lower bounds in decreasing order , we hace . An entirely analogous argument will give an upper bound, yielding
(4)
For those interested in the derivation, read on the in the following optional section:
Derivation of the Mathias–Li Bounds
Since and are diagonal, the eigenvectors of the pair are just the standard basis vectors, the th of which we will denote . The trick will be to use the max-min characterization (1) with the subspace spanned by some collection of basis vectors . Churning through a couple inequalities in quick fashion,23See pg. 17 of the Mathias and Li report. we obtain
Here, denotes the convex hull. Since this holds for every set of indices , it in particular holds for the set of indices which makes the largest. Thus, .
How to Use Mathias–Li’s Perturbation Theory
The eigenangle perturbation bound (4) can be instantiated in a variety of ways. We briefly sketch two. The first is to bound by its minimum over all , which then gives a bound on (and )
Plugging into (4) and simplifying gives
(5)
This improves on Stewart’s bound (3) by replacing the Crawford number by ; as Mathias and Li show is always smaller than or equal to and can be much much smaller.24Recall that Mathias and Li’s bound first requires us to do a change of variables where and both become diagonal, which can increase by a factor of . Thus, for an apples-to-apples comparison with Stewart’s theory where and are non-diagonal, (5) should be interpreted as .
For the second instantiation (4), we recognize that if an eigenangle is sufficiently well-separated from other eigenangles (relative to the size of the perturbation and ), then we have and . (The precise instantiation of “sufficiently well-separated” requires some tedious algebra; if you’re interested, see Footnote 7 in Mathias and Li’s paper.25You may also be interested in Corollary 2.2 in this preprint by myself and coauthors.) Under this separation condition, (4) then reduces to
(6)
This result improves on Stewart’s result (4) by even more, since we have now replaced the Crawford number by for a sufficiently small perturbation. In fact, a result of this form is nearly as good as one could hope for.26Specifically, the condition number of the eigenangle is , so we know for sufficiently small perturbations we have and is the smallest number for which such a relation holds. Mathias and Li’s theory allows for a statement of this form to be made rigorous for a finite-size perturbation. Again, the only small deficit is the additional factor of “” from the change of variables to diagonal form.
The Elegant Geometry of Generalized Eigenvalue Perturbation Theory
As I said at the start of this post, what fascinates me about this generalized eigenvalue perturbation is the beautiful and elegant geometry. When I saw it for the first time, it felt like a magic trick: a definite generalized eigenvalue problem with real eigenvalues was transformed by sleight of hand into a geometry problem on the complex plane, with solutions involving just a little high school geometry and trigonometry. Upon studying the theory, I began to appreciate it for a different reason. Upon closer examination, the magic trick was revealed to be a sequence of deductions, each logically following naturally from the last. To the pioneers of this subject—Stewart, Sun, Mathias, Li, and others—this sequence of logical deductions was not preordained, and their discovery of this theory doubtlessly required careful thought and leaps of insight. Now that this theory has been discovered, however, we get the benefit of retrospection, and can retell a narrative of this theory where each step follows naturally from the last. When told this way, one almost imagines being able to develop this theory by oneself, where at each stage we appeal to some notion of mathematical elegance (e.g., by treating and symmetrically) or by applying a standard trick (e.g., identifying a pair with the complex number ). Since this theory took several decades to fall into place, we should not let this storytelling exercise fool us into thinking the prospective act of developing a new theory will be as straightforward and linear as this retelling, pruned of dead ends and halts in progress, might suggest.
That said, I do think the development of the perturbation theory of the generalized eigenvalue problem does have a lesson for those of us who seek to develop mathematical theories: be guided by mathematical elegance. At several points in the development of the perturbation theory, we obtained great gains by treating quantities which play a symmetric role in the problem symmetrically in the theory or by treating a pair of real numbers as a complex number and asking how to interpret that complex number. My hope is that this perturbation theory serves as a good example for how letting oneself be guided by intuition, a small array of standard tricks, and a search for elegance can lead one to conceptualize a problem in the right way which leads (after a considerable amount of effort and a few lucky breaks) to a natural solution.
In this post, I want to answer a simple question: how can randomness help in solving a deterministic (non-random) problem?
Let’s start by defining some terminology. An algorithm is just a precisely defined procedure to solve a problem. For example, one algorithm to compute the integral of a function on the interval is to pick 100 equispaced points on this interval and output the Riemann sum . A randomized algorithm is simply an algorithm which uses, in some form, randomly chosen quantities. For instance, a randomized algorithm1This randomized algorithm is an example of the strategy of Monte Carlo integration, which has proved particularly effective at computing integrals of functions on high-dimensional spaces. for computing integrals would be to pick 100 random points uniformly distributed in the interval and output the average . To help to distinguish, an algorithm which does not use randomness is called a deterministic algorithm.
This may seem puzzling and even paradoxical. There is no randomness in, say, a system of linear equations, so why should introducing randomness help solve such a problem? It can seem very unintuitive that leaving a decision in an algorithm up to chance would be better than making an informed (and non-random) choice. Why do randomized algorithms work so well; what is the randomness actually buying us?
A partial answer to this question is that it can be very costly to choose the best possible option at runtime for an algorithm. Often, a random choice is “good enough”.
A Case Study: Quicksort
Consider the example of quicksort. Given a list of integers to sort in increasing order, quicksort works by selecting an element to be the pivot and then divides the elements of the list into groups larger and smaller than the pivot. One then recurses on the two groups, ending up with a sorted list.
The best choice of pivot is the median of the list, so one might naturally think that one should use the median as the pivot for quicksort. The problem with this reasoning is that finding the median is time-consuming; even using the fastest possible median-finding algorithm,3There is an algorithm guaranteed to find the median in time, which is asymptotically fast as this problem could possibly be solved since any median-finding algorithm must in general look at the entire list.quicksort with exact median pivot selection isn’t very quick. However, one can show quicksort with a pivot selected (uniformly) at random achieves the same expected runtime as quicksort with optimal pivot selection and is much faster in practice.4In fact, is the fastest possible runtime for any comparison sorting algorithm.
But perhaps we have given up on fast deterministic pivot selection in quicksort too soon. What if we just pick the first entry of the list as the pivot? This strategy usually works fairly well too, but it runs into an unfortunate shortfall: if one feeds in a sorted list, the first-element pivot selection is terrible, resulting in a algorithm. If one feeds in a list in a random order, the first-element pivot selection has a expected runtime,5Another way of implementing randomized quicksort is to first randomize the list and then always pick the first entry as the pivot. This is fully equivalent to using quicksort with random pivot selection in the given ordering. Note that randomizing the order of the list before its sorted is still a randomized algorithm because, naturally, randomness is needed to randomize order. but for certain specific orderings (in this case, e.g., the sorted ordering) the runtime is a disappointing . The first-element pivot selection is particularly bad since its nemesis ordering is the common-in-practice already-sorted ordering. But other simple deterministic pivot selections are equally bad. If one selects, for instance, the pivot to be the entry in the position , then we can still come up with an ordering of the input list that makes the algorithm run in time . And because the algorithm is deterministic, it runs in time every time with such-ordered inputs. If the lists we need to sort in our code just happen to be bad for our deterministic pivot selection strategy, quicksort will be slow every time.
We typically analyze algorithms based on their worst-case performance. And this can often be unfair to algorithms. Some algorithms work excellently in practice but are horribly slow on manufactured examples which never occur in “real life”.6The simplex algorithm is an example of algorithm which is often highly effective in practice, but can take a huge amount of time on some pathological cases. But average-case analysis of algorithms, where one measures the average performance of an algorithm over a distribution of inputs, can be equally misleading. If one were swayed by the average-case analysis of the previous section for quicksort with first-element pivot selection, one would say that this should be an effective pivot selection in practice. But sorting an already-sorted or nearly-sorted list occurs all the time in practice: how many times do programmers read in a sorted list from a data source and sort it again just to be safe? In real life, we don’t encounter random inputs to our algorithms: we encounter a very specific distribution of inputs specific to the application we use the algorithm in. An algorithm with excellent average-case runtime analysis is of little use to me if it is consistently and extremely slow every time I run it on my input data on the problem I’m working on. This discussion isn’t meant to disparage average-case analysis (indeed, very often the “random input” model is not too far off), but to explain why worst-case guarantees for algorithms can still be important.
To summarize, often we have a situation where we have an algorithm which makes some choice (e.g. pivot selection). A particular choice usually works well for a randomly chosen input, but can be stymied by certain specific inputs. However, we don’t get to pick which inputs we receive, and it is fully possible the user will always enter inputs which are bad for our algorithms. Here is where randomized algorithms come in. Since we are unable to randomize the input, we instead randomize the algorithm.
A randomized algorithm can be seen as a random selection from a collection of deterministic algorithms. Each individual deterministic algorithm may be confounded by an input, but most algorithms in the collection will do well on any given input. Thus, by picking a random algorithm from our collection, the probability of poor algorithmic performance is small. And if we run our randomized algorithm on an input and it happens to do poorly by chance, if we run it again it is unlikely to do so; there isn’t a specific input we can create which consistently confounds our randomized algorithm.
The Game Theory Framework
Let’s think about algorithm design as a game. The game has two players: you and an opponent. The game is played by solving a certain algorithmic problem, and the game has a score which quantifies how effectively the problem was solved. For example, the score might be the runtime of the algorithm, the amount of space required, or the error of a computed quantity. For simplicity of discussion, let’s use runtime as an example for this discussion. You have to pay your opponent a dollar for every second of runtime, so you want to have the runtime be as low as possible.7In this scenario, we consider algorithms which always produce the correct answer to the problem, and it is only a matter of how long it takes to do so. In the field of algorithms, randomized algorithms of this type are referred to as Las Vegas algorithms. Algorithms which can also give the wrong answer with some (hopefully small) probability are referred to as Monte Carlo algorithms.
Each player makes one move. Your move is to present an algorithm which solves the problem. Your opponent’s move is to provide an input to your algorithm. Your algorithm is then run on your opponents input and you pay your opponent a dollar for every second of runtime.
This setup casts algorithm design as a two-person, zero-sum game. It is a feature of such games that a player’s optimal strategy is often mixed, picking a move at random subject to some probability distribution over all possible moves.
To see why this is the case, let’s consider a classic and very simple game: rock paper scissors. If I use a deterministic strategy, then I am forced to pick one choice, say rock, and throw it every time. But then my savvy opponent could pick paper and be assured victory. By randomizing my strategy and selecting between rock, paper, and scissors (uniformly) at random, I improve my odds to winning a third of the time, losing a third of the time, and drawing a third of the time. Further, there is no way my opponent can improve their luck by adopting a different strategy. This strategy is referred to as minimax optimal: among all (mixed) strategies I could adopt, this strategy has the best performance over all strategies provided my opponent always counters my strategy with the best response strategy they can find.
Algorithm design is totally analogous. For the pivot selection problem, if I pick a the fixed strategy of choosing the first entry to be the pivot (analogous to always picking rock), then my opponent could always give me a sorted list (analogous to always picking paper) and I would always lose. My randomizing my pivot selection, my opponent could input the list in whatever order they choose and my pivot selection will always have the excellent (expected) runtime characteristic of quicksort. In fact, randomized pivot selection is the minimax optimal pivot selection strategy (assuming pivot selection is non-adaptive in that we don’t choose the pivot based on the values in the list).8This is not to say that quicksort with randomized pivot selection is necessarily the minimax optimal sorting algorithm, but to say that once we have fixed quicksort as our sorting algorithm, randomized pivot selection is the minimax optimal non-adaptive pivot selection strategy for quicksort.
How Much Does Randomness Buy?
Hopefully, now, the utility of randomness is less opaque. Randomization, in effect, allows an algorithm designer to trade algorithm which runs fast for most inputs all of the time for an algorithm which runs fast for all inputs most of the time. They do this by introducing randomized decision-making to hedge against particular bad inputs which could confound their algorithm.
Randomness, of course, is not a panacea. Randomness does not allow us to solve every algorithmic problem arbitrarily fast. But how can we quantify this? Are there algorithmic speed limits for computational problems for which no algorithm, randomized or not, can exceed?
The game theoretic framework for randomized algorithms can shed light on this question. Let us return to the framing of last section where you choose an algorithm , your opponent chooses an input , and you pay your opponent a dollar for every second of runtime. Since this cost depends on the algorithm and the input, we can denote the cost .
Suppose you devise a randomized algorithm for this task, which can be interpreted as selecting an algorithm from a probability distribution over the class of all algorithms which solve the problem. (Less formally, we assign each possible algorithm a probability of picking it and pick one subject to these probabilities.) Once your opponent sees your randomized algorithm (equivalently, the distribution ), they can come up with the on-average slowest possible input and present it to your algorithm.
But now let’s switch places to see things from your opponent. Suppose they choose their own strategy of randomly selecting an input from among all inputs with some distribution . Once you see the distribution , you can come up with the best possible deterministic algorithm to counter their strategy.
The next part gets a bit algebraic. Suppose now we apply your randomized algorithm against your opponents strategy. Then, your randomized algorithm could only take longer on average than because, by construction, is the fastest possible algorithm against the input distribution . Symbolically, we have
(1)
Here, and denote the average (expected) value of a random quantity when an input is drawn randomly with distribution or an algorithm is drawn randomly with distribution . But we know that is the slowest input for our randomized algorithm, so, on average, our randomized algorithm will take longer on worst-case input then a random input from . In symbols,
In words, the average performance of any randomized algorithm on its worst-case input can be no better than the average performance of the best possible deterministic algorithm for adistribution of inputs.10In fact, there is a strengthening of Yao’s minimax principle. That Eqs. (1) and (2) are equalities when and are taken to be the minimax optimal randomized algorithm and input distribution, respectively. Specifically, assuming the cost is a natural number and we restrict to a finite class of potential inputs, This nontrivial fact is a consequence of the full power of the von Neumann’s minimax theorem, which itself is a consequence of strong duality in linear programming. My thanks go to Professor Leonard Schulman for teaching me this result.
This, in effect, allows the algorithm analyst seeking to prove “no randomized algorithm can do better than this” to trade randomness in the algorithm to randomness in the input in their analysis. This is a great benefit because randomness in an algorithm can be used in arbitrarily complicated ways, whereas random inputs can be much easier to understand. To prove any randomized algorithm takes at least cost to solve a problem, the algorithm analyst can find a distribution on which every deterministic algorithm takes at least cost . Note that Yao’s minimax principle is an analytical tool, not an algorithmic tool. Yao’s principle establishes speed limits on the best possible randomized algorithm: it does not imply that one can just use deterministic algorithms and assume or make the input to be random.
There is a fundamental question concerning the power of randomized algorithms not answered by Yao’s principle that is worth considering: how much better can randomized algorithms be than deterministic ones? Could infeasible problems for deterministic computations be solved tractably by randomized algorithms?
Questions such as these are considered in the field of computational complexity theory. In this context, one can think of a problem as tractably solvable if it can be solved in polynomial time—that is, in an amount of time proportional to a polynomial function of the size of the input. Very roughly, we call the class of all such problems to be . If one in addition allows randomness, we call this class of problems.11More precisely, is the class of problems solvable in polynomial time by a Monte Carlo randomized algorithm. The class of problems solvable in polynomial time by a Las Vegas randomized algorithm, which we’ve focused on for the duration of this article, is a possibly smaller class called .
It has widely been conjectured that : all problems that can be tractably solved with randomness can tractably be solved without. There is some convincing evidence for this belief. Thus, provided this conjecture turns out to be true, randomness can give us reductions in operation counts by a polynomial amount in the input size for problems already in , but they cannot efficiently solve -hard computational problems like the traveling salesman problem.12Assuming the also quite believable conjecture that .
So let’s return to the question which is also this article’s title: why randomized algorithms? Because randomized algorithms are often faster. Why, intuitively, is this the case? Randomization can us to upgrade simple algorithms that are great for most inputs to randomized algorithms which are great most of the time for all inputs. How much can randomization buy? A randomized algorithm on its worst input can be no better than a deterministic algorithm on a worst-case distribution. Assuming a widely believed and theoretically supported, but not yet proven, conjecture, randomness can’t make intractable problems into tractable ones. Still, there is great empirical evidence that randomness can be an immensely effective algorithm design tool in practice, including computational math and science problems like trace estimation and solving Laplacian linear systems.
A singular value decomposition of an matrix is a factorization of the form , where and are square, orthogonal matrices and is a diagonal matrix with th entry .1Everything carries over essentially unchanged for complex-valued matrices with and being unitary matrices and every being replaced by for the Hermitian transpose. The diagonal entries of are referred to as the singular values of and are conventionally ordered . The columns of the matrices and are referred to as the right- and left- singular vectors of and satisfy the relations and .
One can obtain the singular values and right and left singular vectors of from the eigenvalues and eigenvectors of and . This follows from the calculations and . In other words, the nonzero singular values of are the square roots of the nonzero eigenvalues of and . If one merely solves one of these problems, computing along with or , one can obtain the other matrix or by computing or . (These formulas are valid for invertible square matrices , but similar formulas hold for singular or rectangular to compute the singular vectors with nonzero singular values.)
This approach is often unundesirable for several reasons. Here are a few I’m aware of:
Accuracy: Roughly speaking, in double-precision arithmetic, accurate stable numerical methods can resolve differences on the order of 16 orders of magnitude. This means an accurately computed SVD of can resolve the roughly 16 orders of magnitude of decaying singular values, with singular values smaller than that difficult to compute accurately. By computing , we square all of our singular values, so resolving 16 orders of magnitude of the eigenvalues of means we only resolve 8 orders of magnitude of the singular values of .2Relatedly, the two-norm condition number of is twice that of . The dynamic range of our numerical computations has been cut in half!
Loss of orthogonality: While and are valid formulas in exact arithmetic, they fair poorly when implemented numerically. Specifically, the numerically computed values and may not be orthogonal matrices with, for example, not even close to the identity matrix. One can, of course, orthogonalize the computed or , but this doesn’t fix the underlying problem that or have not been computed accurately.
Loss of structure: If possesses additional structure (e.g. sparsity), this structure may be lost or reduced by computing the product .
There are times where these problems are insignificant and this approach is sensible: we shall return to this point in a bit. However, these problems should disqualify this approach from being the de facto way we reduce SVD computation to a symmetric eigenvalue problem. This is especially true since we have a better way.
The better way is by constructing the so-called Hermitian dilation4As Stewart and Sun detail in Section 4 of Chapter 1 of their monograph Matrix Perturbation Theory, the connections between the Hermitian dilation and the SVD go back to the discovery of the SVD itself, as it is used in Jordan’s construction of the SVD in 1874. (The SVD was also independently discovered by Beltrami the year previous.) Stewart and Sun refer to this matrix as the Jordan-Wiedlant matrix associated with , as they attribute the widespread use of the matrix today to the work of Wiedlant. We shall stick to the term Hermitian dilation to refer to this matrix. of , which is defined to be the matrix
(1)
One can show that the nonzero eigenvalues of are precisely plus-or-minus the singular values of . More specifically, we have
(2)
All of the remaining eigenvalues of not of this form are zero.5This follows by noting and thus for account for all the nonzero eigenvalues of . Thus, the singular value decomposition of is entirely encoded in the eigenvalue decomposition of .
This approach of using the Hermitian dilation to compute the SVD of fixes all the issues identified with the “” approach. We are able to accurately resolve a full 16 orders of magnitude of singular values. The computed singular vectors are accurate and numerically orthogonal provided we use an accurate method for the symmetric eigenvalue problem. The Hermitian dilation preserves important structural characteristics in like sparsity. For purposes of theoretical analysis, the mapping is linear.6The linearity of the Hermitian dilation gives direct extensions of most results about the symmetric eigenvalues to singular values; see Exercise 22.
Often one can work with the Hermitian dilation only implicitly: the matrix need not actually be stored in memory with all its extra zeros. The programmer designs and implements an algorithm with in mind, but deals with the matrix directly for their computations. In a pinch, however, forming directly in software and utilizing symmetric eigenvalue routines directly is often not too much less efficient than a dedicated SVD routine and can cut down on programmer effort significantly.
As with all things in life, there’s no free lunch here. There are a couple of downsides to the Hermitian dilation approach. First, is, except for the trivial case , an indefinite symmetric matrix. By constast, and are positive semidefinite, which can be helpful in some contexts.7This is relevant if, say, we want to find the small singular values by inverse iteration. Positive definite linear systems are easier to solve by either direct or iterative methods. Further, if (respectively, ), then (respectively, ) is tiny compared to , so it might be considerably cheaper to compute an eigenvalue decomposition of (or ) than .
Despite the somewhat salacious title of this article, the and Hermitian dilation approaches both have their role, and the purpose of this article is not to say the approach should be thrown in the dustbin. However, in my experience, I frequently hear the approach stated as the definitive way of converting an SVD into an eigenvalue problem, with the Hermitian dilation approach not even mentioned. This, in my opinion, is backwards. For accuracy reasons alone, the Hermitian dilation should be the go-to tool for turning SVDs into symmetric eigenvalue problems, with the approach only used when the problem is known to have singular values which don’t span many orders of magnitude or is tall and skinny and the computational cost savings of the approach are vital.
be a (real) random variable and
a convex function such that both
and
are defined. Then
.
 , there exists a slope
, there exists a slope  such that
such that  for all
for all  . Invoke this result at
. Invoke this result at  and
and  and take expectations to conclude that
and take expectations to conclude that![\[\mathbb{E}[m(X - \mathbb{E}X) + f(\mathbb{E}X)] = f(\mathbb{E}X) \le \mathbb{E} [f(X)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2eb518dc5399785b559f7e3d9684fdf9_l3.png "Rendered by QuickLaTeX.com")
 for two numbers
for two numbers  and
and  . This may hard to do directly. With the interpolation method, I first construct a family of numbers
. This may hard to do directly. With the interpolation method, I first construct a family of numbers  ,
,  , such that
, such that  and
and  and show that
and show that  is (weakly) increasing in
is (weakly) increasing in  . This is typically accomplished by showing the derivative is nonnegative:
. This is typically accomplished by showing the derivative is nonnegative:![\[\frac{d}{dt} A_t \ge 0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c22b7fde162a006b2090375c734d168e_l3.png "Rendered by QuickLaTeX.com")
) and let
for some
for any Gaussian random variable
. Then
![\[f(0) \le \mathbb{E} [f(X)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1f17b24cdabfb933b0cc38f8f5ad867f_l3.png "Rendered by QuickLaTeX.com")
![\[A_t = \mathbb{E} [ f(X_t) ],\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5f3b73e5d58b2d3f23313503392f7487_l3.png "Rendered by QuickLaTeX.com")
 starts with no randomness and
starts with no randomness and  is our standard Gaussian. To interpolate between these extremes, we increase the variance linearly from
is our standard Gaussian. To interpolate between these extremes, we increase the variance linearly from  to
to ![\[A_t = \mathbb{E} [ f(X_t)] \quad \text{where $X_t\sim\mathcal{N}(0,t)$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-92aec0d0314cfb18b1400559a7b05458_l3.png "Rendered by QuickLaTeX.com")
 denotes a Gaussian random variable with zero mean and variance
denotes a Gaussian random variable with zero mean and variance  .
. denote a small parameter which we will later send to zero. For us, the key fact will be that a
denote a small parameter which we will later send to zero. For us, the key fact will be that a  can be realized as a sum of independent
can be realized as a sum of independent  and
and  random variables. Therefore, we write
random variables. Therefore, we write![\[X_{t+\delta} = X_t + \Delta \quad \text{where $\Delta \sim \mathcal{N}(0,\delta)$ is independent of $X_t$.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-39ac434efed935085da4cf9b8f818afa_l3.png "Rendered by QuickLaTeX.com")
 by using Taylor’s formula
by using Taylor’s formula![\[f(X_t+\Delta) = f(X_t) + f'(X_t)\Delta + \frac{1}{2} f''(X_t) \Delta^2 + \frac{1}{6} f'''(\xi) \Delta^3, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d7e58c5f0f88658410b4a5695f183ffc_l3.png "Rendered by QuickLaTeX.com")
 lies between
lies between  and
and  . Now, take expectations,
. Now, take expectations, ![\[\mathbb{E}[ f(X_t+\Delta)]=\mathbb{E}[f(X_t)] + \mathbb{E}[f'(X_t)\Delta] + \frac{1}{2} \mathbb{E}[f''(X_t)] \mathbb{E}[\Delta^2] + \underbrace{\frac{1}{6} \mathbb{E}[f'''(\xi) \Delta^3]}_{:=\mathrm{Rem}(\delta)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2254d2d0eaaf30aa3fef139ee401cc4d_l3.png "Rendered by QuickLaTeX.com")
 has mean zero and variance
has mean zero and variance  so this gives
so this gives![\[\mathbb{E} [f(X_t+\Delta)]=\mathbb{E}[f(X_t)] + \delta \frac{1}{2} \mathbb{E}[f''(X_t)] + \mathrm{Rem}(\delta).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-faedc3b7ac6aee830b116b6dc50b9e5b_l3.png "Rendered by QuickLaTeX.com")
 vanishes as
vanishes as  . Thus, we can rearrange this expression to compute the derivative:
. Thus, we can rearrange this expression to compute the derivative:![\[\frac{d}{dt} A_t = \lim_{\delta \downarrow 0} \frac{\mathbb{E} f(X_t+\Delta)-\mathbb{E}[f(X_t)]}{\delta} = \lim_{\delta \downarrow 0} \frac{1}{2} \mathbb{E}[f''(X_t)] + \frac{\mathrm{Rem}(\delta)}{\delta} = \frac{1}{2} \mathbb{E}[f''(X_t)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fe0e1500ae627522def30f418f91f38b_l3.png "Rendered by QuickLaTeX.com")
 for every
for every  . Therefore,
. Therefore,![\[\frac{d}{dt} A_t \ge 0 \quad \text{for all } t\in [0,1].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7ac1565d61bc801156b3ff56ff95ceb8_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} f(X) = f(0) + \frac{1}{2} \int_0^1 \mathbb{E} [f''(X_t)] \, dt.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4eb33e5cc30d654146cfb21e9269eebe_l3.png "Rendered by QuickLaTeX.com")
 is
is  -smooth
-smooth![\[f''(x) \le \beta \quad \text{for every } x \in \real,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c94168730ce96b43c38cc0b338afbae5_l3.png "Rendered by QuickLaTeX.com")
![\[f(0) \le \mathbb{E} f(X) \le f(0) + \frac{1}{2}\beta.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fbcdd9029364fb94f1a7e669e7327e87_l3.png "Rendered by QuickLaTeX.com")
 . Let’s do this. As an exercise, you can verify that our technical regularity condition implies
. Let’s do this. As an exercise, you can verify that our technical regularity condition implies  . Thus, by Hölder’s inequality and setting
. Thus, by Hölder’s inequality and setting  to be
to be  ‘s Hölder conjugate (
‘s Hölder conjugate ( ), we obtain
), we obtain![\[\frac{|\mathrm{Rem}(\delta)|}{\delta} = \frac{|\mathbb{E}[f'''(\xi) \Delta^3]|}{6\delta} \le \frac{(|\mathbb{E} |f'''(\xi)|^p)^{1/p}| (\mathbb{E} |\Delta|^{3q})^{1/q}}{6\delta}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-adf44eea269d8b70504625e4664b1ccc_l3.png "Rendered by QuickLaTeX.com")
 where
where  is a function of
is a function of  as
as  .
. , defined for each
, defined for each  . Rather than treating these quantities as independent, we can think of them as a collective, comprising a random function
. Rather than treating these quantities as independent, we can think of them as a collective, comprising a random function  known as a Brownian motion.
known as a Brownian motion.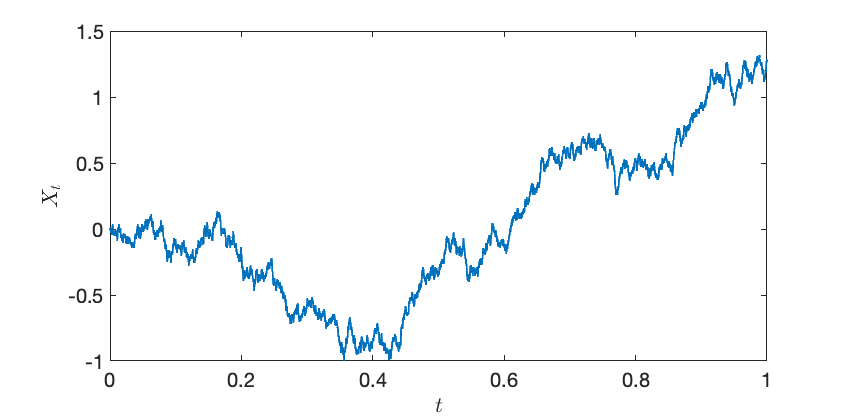
 and a Gaussian with mean
and a Gaussian with mean ![\[df = f'(x) \, dx\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4814213cc8461805f3f382303ae1fd44_l3.png "Rendered by QuickLaTeX.com")
 of a Brownian motion, the analog is Itô’s formula
of a Brownian motion, the analog is Itô’s formula![\[df = f'(X_t) \, dX_t + \frac{1}{2} f''(X_t) \, dt.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-80e8633a48870a6fe8548cae58fc3651_l3.png "Rendered by QuickLaTeX.com")
 over a time interval
over a time interval  ,
,  is a random variable with mean
is a random variable with mean  :
:![\[f(\mathbb{E}Y) \le \mathbb{E}[f(Y)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ac997c472db630b535613f75c391df96_l3.png "Rendered by QuickLaTeX.com")
 and
and  denote the cumulative distribution functions of
denote the cumulative distribution functions of ![\[g(X) := \inf \{ \alpha \in \real : F_Y(\alpha) \ge F_X(X) \}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6bd5ab53ed0082ed885e7f69ba1736ad_l3.png "Rendered by QuickLaTeX.com")
 and
and ![\[A_t \stackrel{?}{=} \mathbb{E} [f(g(X_t))].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3e35478dad25431a7564f48d438bbc1c_l3.png "Rendered by QuickLaTeX.com")
![A_0 = \mathbb{E}[f(g(0))]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4ee3e7fbbdfe560f22d49739bc7e83fe_l3.png "Rendered by QuickLaTeX.com") does not even equal to
does not even equal to ![\mathbb{E} [f(Y)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-85c0709ac3c82a809fa5e7afc9c64b8d_l3.png "Rendered by QuickLaTeX.com") ! Instead, we must define
! Instead, we must define![\[A_t = \mathbb{E} [f(\mathbb{E}[g(X_1) \mid X_t])].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e14b936c760d07f6b598dfdb2e41bc00_l3.png "Rendered by QuickLaTeX.com")
 conditional on the Brownian motion
conditional on the Brownian motion ![\[\frac{d}{dt} A_t \ge 0 \quad \text{for all }t\in [0,1].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fe036e08c1885785cea732f2f8c1b510_l3.png "Rendered by QuickLaTeX.com")
are “somewhat dependent”, for which functions does the multivariate Jensen’s inequality
)
![\[f(\mathbb{E} Y_1,\ldots,\mathbb{E}Y_n) \le \mathbb{E} [f(Y_1,\ldots,Y_n)] \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-43d7e890fa9211f1b4e6e83f2bc1ce08_l3.png "Rendered by QuickLaTeX.com")
![\[\text{($\star$) holds if $f$ is convex in each coordinate, separately.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7473686cff7032897db3fc21073ffc50_l3.png "Rendered by QuickLaTeX.com")
 of Gaussian random variables
of Gaussian random variables  . We will use the covariance matrix
. We will use the covariance matrix  of the Gaussian random variables
of the Gaussian random variables if and only if
is the Hessian matrix at
denotes the entrywise product of matrices.
![\[f(\mathbb{E}g_1(X_1),\ldots,\mathbb{E}g_n(X_n)) \le \mathbb{E} [f(g(X_1),\ldots,g(X_n))]\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-37d1c91ba37e25c20d03265695b28944_l3.png "Rendered by QuickLaTeX.com")
![\[\Sigma \circ \nabla^2 f(x) \text{ is positive semidefinite} \quad \text{for all $x \in \real^n$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-412da3a61a850bd039a68703b09afbd6_l3.png "Rendered by QuickLaTeX.com")
 are equal (and variance one),
are equal (and variance one),  for all
for all ![\[\Sigma \circ \nabla^2 f(x) = \mathrm{diag} \left( \frac{\partial^2 f}{\partial x_i^2} : i=1,\ldots,n \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e0c6fd2476ffdbfa9f210e476fc0c313_l3.png "Rendered by QuickLaTeX.com")
 : this is precisely the condition for
: this is precisely the condition for  via the matrix–vector product operation
via the matrix–vector product operation  , estimate its trace
, estimate its trace  .
.![\[\hat{\tr} = \frac{1}{m} \sum_{i=1}^m \omega_i^* A \omega_i. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d7de978db4f57321a96c53944bebc189_l3.png "Rendered by QuickLaTeX.com")
 are random vectors, usually chosen to be
are random vectors, usually chosen to be  denotes the
denotes the  .
.![\[\mathbb{E} [\omega_i\omega_i^*] = I, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d962300e9943c0618c633bfeefb289f0_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} [\hat{\tr}] = \tr A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-210efada631bdca25b3196a9c9aae00d_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} [\hat{\tr}] = \mathbb{E} \left[ \frac{1}{m} \sum_{i=1}^m \omega_i^*A\omega_i \right] = \frac{1}{m} \sum_{i=1}^m \mathbb{E} \left[ \omega_i^* A \omega_i\right]. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5cb7027c877f59aae291b08b47d47966_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E} [\hat{\tr}] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-05f499d7b3aeb0ed594301b2ac751441_l3.png "Rendered by QuickLaTeX.com") , it is sufficient to prove that
, it is sufficient to prove that ![\mathbb{E} \left[\omega_i^*A\omega_i\right] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-13de1504d7bf46d1956279a7e4d28e01_l3.png "Rendered by QuickLaTeX.com") for each
for each  .
. matrix, then a number equals its trace,
matrix, then a number equals its trace,  . The second trick is the
. The second trick is the  matrix
matrix  and a
and a  matrix
matrix  , we have
, we have  . The cyclic property
. The cyclic property ![\[\tr[BCD] = \tr[(BC)D] = \tr[D(BC)] = \tr[DBC].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-155ce79a5ef10f252cd9517740583182_l3.png "Rendered by QuickLaTeX.com")
![\[\tr [BCD] \ne \tr[CBD] \quad \text{in general}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2b72ed0e84a060cf970349f61aed68ee_l3.png "Rendered by QuickLaTeX.com")
 as beads on a closed loop of string. One can move the last bead
as beads on a closed loop of string. One can move the last bead  to the front of the other two,
to the front of the other two, ![\tr [BCD] = \tr[DBC]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-af77d563ff63b634047619d25a5a83bf_l3.png "Rendered by QuickLaTeX.com") , but not interchange two beads,
, but not interchange two beads, ![\tr[BCD] \ne \tr[CBD]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-236f232e9f446e824ab55cf3f19b4895_l3.png "Rendered by QuickLaTeX.com") .
.![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[A\omega_i\omega_i^*\right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9187ce141ddc88ae2c5f717c27571141_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[A\omega_i\omega_i^*\right] = \tr(A \mathbb{E}[\omega_i\omega_i^*] ).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a260629d9c41472ea9b71c3bb10a2a1_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \tr(A \mathbb{E}[\omega_i\omega_i^*] ) = \tr(A\cdot I) = \tr A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c1c09245d42d16b1a05566cbe1e27664_l3.png "Rendered by QuickLaTeX.com")
 ‘s are isotropic (3) and now assume that
‘s are isotropic (3) and now assume that ![\[\Var(\hat{\tr}) = \frac{1}{m^2} \sum_{i=1}^m \Var(\omega_i^*A\omega_i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4401469db02c371beb1b1bbaa5de731f_l3.png "Rendered by QuickLaTeX.com")
 , we then get
, we then get![\[\Var(\hat{\tr}) = \frac{1}{m} \Var(\omega^*A\omega).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-40b9400b57ed2e445fc0a0813ed61699_l3.png "Rendered by QuickLaTeX.com")
 , which is characteristic of
, which is characteristic of  is unbiased (i.e, (3)), this means that the mean square error decays like
is unbiased (i.e, (3)), this means that the mean square error decays like ![\[\left| \hat{\tr} - \tr A \right| \lessapprox \frac{\mathrm{const}}{\sqrt{m}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e19b6a62552344261d535991945baf0c_l3.png "Rendered by QuickLaTeX.com")
 , I must do
, I must do  the work!
the work! and (root-mean-square) error at rates
and (root-mean-square) error at rates ![\[\Var(\hat{\tr}_{\text{H++ or X}}) \le \frac{\mathrm{const}}{m^2},\quad \left| \hat{\tr}_{\text{H++ or X}} - \tr A \right| \lessapprox \frac{\mathrm{const}}{m}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-04f981bb20ddfeb465d879e9ef440854_l3.png "Rendered by QuickLaTeX.com")
 that makes the variance of the single–sample estimate
that makes the variance of the single–sample estimate  as low as possible. In this section, we will provide several explicit formulas for the variance of the Girard–Hutchinson estimator. Derivations of these formulas will appear at the end of this post. These variance formulas help illuminate the benefits and drawbacks of different test vector distributions.
as low as possible. In this section, we will provide several explicit formulas for the variance of the Girard–Hutchinson estimator. Derivations of these formulas will appear at the end of this post. These variance formulas help illuminate the benefits and drawbacks of different test vector distributions. we use
we use  and
and  to denote the real and imaginary parts. The variance of a random complex number
to denote the real and imaginary parts. The variance of a random complex number  is
is![\[\Var(z) := \mathbb{E} |z - \mathbb{E} z|^2 = \Var(\Re z) + \Var(\Im z).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a02aee32ffbeff14f4a44921d3324126_l3.png "Rendered by QuickLaTeX.com")
![\[\left\|A\right\|_{\rm F}^2 = \sum_{i,j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5cb12b8a023d04d9bbfff1e1cc685bb3_l3.png "Rendered by QuickLaTeX.com")
 , we have
, we have![\[\left\|A\right\|_{\rm F}^2 = \sum_{i=1}^n \lambda_i^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a67a31b37b18f53f5d060e94c896ebc6_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  denotes the
denotes the  rather than the conjugate transpose
rather than the conjugate transpose  is a number, it is equal to its own transpose:
is a number, it is equal to its own transpose:![\[\omega^\top A \omega = (\omega^\top A \omega)^\top = \omega^\top A^\top \omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-719b03091ae9961865f7c26c73b089cb_l3.png "Rendered by QuickLaTeX.com")
![\[\omega^\top A\omega = \frac{\omega^\top A \omega + \omega^\top A^\top \omega}{2} = \omega^\top \left( \frac{A + A^\top}{2} \right)\omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-26830e0938226dfafe0012340b3edc4c_l3.png "Rendered by QuickLaTeX.com")
 .
. .
.![\[\Var(\omega^\top A\omega) = 2 \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-68d0725a0be66f7bd471887707dc00e9_l3.png "Rendered by QuickLaTeX.com")

![\[\Var(\omega^\top A \omega) = 2\sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d39bacfe5e266b7e6a389b49a2ef7ab4_l3.png "Rendered by QuickLaTeX.com")
 are uniformly distributed on the real sphere of radius
are uniformly distributed on the real sphere of radius  :
:  .
. ![\[\Var(\omega^\top A\omega) = \frac{2n}{n+2} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n} |\tr A|^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-62d79e990b1371fe7f3b70afd022d359_l3.png "Rendered by QuickLaTeX.com")
![\[A = A^{\rm H} + i A^{\rm SH}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8ec815ab2ed2bfe74d2eab50fc83bbf2_l3.png "Rendered by QuickLaTeX.com")
![\[A^{\rm H} = \frac{A+A^*}{2} ,\quad A^{\rm SH} = \frac{A - A^*}{2i}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1f89c50f79ec3d8662597498c14792b7_l3.png "Rendered by QuickLaTeX.com")
 is skew-Hermitian). Since
is skew-Hermitian). Since  and
and  are both Hermitian, we have
are both Hermitian, we have![\[\Re(\omega^* A\omega) = \omega^* A^{\rm H} \omega, \quad \Im (\omega^* A \omega) = \omega^* A^{\rm SH} \omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-222e3ce55f034ead06188b52f756dbf4_l3.png "Rendered by QuickLaTeX.com")
 can be broken into Hermitian and skew-Hermitian parts:
can be broken into Hermitian and skew-Hermitian parts:![\[\Var(\omega^* A\omega) = \Var(\omega^* A^{\rm H}\omega) + \Var(\omega^* A^{\rm SH}\omega).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-52f0bee30b0c5a3011d04a696be9a63b_l3.png "Rendered by QuickLaTeX.com")
 .
. for
for  standard normal random variables.
standard normal random variables. ![\[\Var(\omega^* A\omega) = \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cc3fa5a76928939dc4e59e4e579719ec_l3.png "Rendered by QuickLaTeX.com")
 .
. ![\[\Var(\omega^* A \omega) = \sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d82497dca5262e5242afb868d5a4fa08_l3.png "Rendered by QuickLaTeX.com")
 .
. ![\[\Var(\omega^* A\omega) = \frac{n}{n+1} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n} |\tr A|^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-23aeee1ea3a346726462faee63b50656_l3.png "Rendered by QuickLaTeX.com")
 are isotropic (2), independent from each other, and have independent entries, then for any fixed real symmetric matrix
are isotropic (2), independent from each other, and have independent entries, then for any fixed real symmetric matrix  .
. -valued and an
-valued and an  -valued test vectors on unified footing, let
-valued test vectors on unified footing, let  denote either
denote either  ) or a complex Hermitian matrix (if
) or a complex Hermitian matrix (if  ). Let a
). Let a  of
of ![\[\text{If $A \in \mathscr{A}$ and $U$ is $\field$-unitary, then $U^*AU \in \mathscr{A}$.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fd95bf4747231110e72c59c18e4b5794_l3.png "Rendered by QuickLaTeX.com")
 is minimized by choosing
is minimized by choosing  .
. , the set of all
, the set of all  , where
, where  is
is  is a diagonal matrix reporting
is a diagonal matrix reporting  has the same distribution as
has the same distribution as  . Therefore,
. Therefore,![\[\Var(\omega^\top A \omega) = \Var(\omega^\top \Lambda \omega) = \Var \left( \sum_{i=1}^n \lambda_i \omega_i^2 \right) = \sum_{i=1}^n \lambda_i^2 \Var(\omega_i^2) = 2\sum_{i=1}^n \lambda_i^2 = 2\left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-88610c0bb2f7d00d6c5b122128b3ecc2_l3.png "Rendered by QuickLaTeX.com")
 . Thus,
. Thus,![\[\Var(\omega^\top A \omega) = \Var(\omega^\top \mathfrak{R}(A) \omega) + \Var(\omega^\top \mathfrak{I}(A) \omega) = 2\left\|\mathfrak{R}(A)\right\|_{\rm F}^2 + 2\left\|\mathfrak{I}(A)\right\|_{\rm F}^2 = 2\left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4a48ec010d732bb8bb81a4e2f62c4a60_l3.png "Rendered by QuickLaTeX.com")
![\[\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega] = \sum_{i,j=1}^n A_{ij} \omega_i\omega_j - \sum_{i=1}^n A_{ii} = \sum_{i\ne j} A_{ij} \omega_i\omega_j + \sum_{i=1}^n A_{ii}(\omega_i^2-1).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2b40ca061457a01f94dd006c31ec31aa_l3.png "Rendered by QuickLaTeX.com")
 for every
for every  can be replaced by two times the sum over
can be replaced by two times the sum over  :
:![\[\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega] = 2\sum_{i< j} A_{ij} \omega_i\omega_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-554778da91da826c0a2eb6a61e95c949_l3.png "Rendered by QuickLaTeX.com")
 are
are ![\[\Var(a_1 X_1+\cdots+a_kX_k) = |a_1|^2 \Var(X_1) + \cdots + |a_k|^2 \Var(X_k)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9bc2039e8f7ba6d806c6c3ebd7a56bd5_l3.png "Rendered by QuickLaTeX.com")
 and numbers
and numbers  . Ergo,
. Ergo,![\begin{align*}\Var(\omega^\top A\omega) &= \Var(\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega]) \\&= \Var\left(\sum_{i< j} 2A_{ij} \omega_i\omega_j\right) \\&= \sum_{i<j} 4 |A_{ij}|^2 \Var(\omega_i\omega_j) \\&= \sum_{i<j} 4 |A_{ij}|^2 \\&= 2 \sum_{i\ne j} |A_{ij}|^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-69e5092cd2e186735dff9ae8f39bf6cb_l3.png "Rendered by QuickLaTeX.com")
 is a uniform random sign, which has variance
is a uniform random sign, which has variance  camels, the second son took
camels, the second son took  camels, the third son
camels, the third son  camels and the wise man took his own camel and went away.
camels and the wise man took his own camel and went away. which has a random length. We want to pick a distribution for which the computation
which has a random length. We want to pick a distribution for which the computation  is easy. Fortunately, we already know such a distribution, the Gaussian distribution, for which we already calculated
is easy. Fortunately, we already know such a distribution, the Gaussian distribution, for which we already calculated  .
.![\[g = \sqrt{\frac{a}{n}} \omega,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a1a3030e4659fe1eb07283cf444cfaa0_l3.png "Rendered by QuickLaTeX.com")
 is the squared length of the Gaussian vector
is the squared length of the Gaussian vector  squared Gaussian random variables, which is known as a
squared Gaussian random variables, which is known as a 
 using the
using the ![\[\Var(g^\top A g) = \mathbb{E}[\Var(g^\top A g \mid a)] + \Var(\mathbb{E}[g^\top A g \mid a]).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-905a9c4d09b2126ed9eed55771631b62_l3.png "Rendered by QuickLaTeX.com")
 and
and ![\mathbb{E}[ \cdot \mid a]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a0e2cfc286402f1e4061fdf9fb3167f_l3.png "Rendered by QuickLaTeX.com") denote the
denote the  and treating
and treating ![\begin{align*}\Var(g^\top A g) &= \mathbb{E}[\Var(a/n \cdot \omega^\top A \omega \mid a)] + \Var(\mathbb{E}[a/n \cdot \omega^\top A \omega \mid a]) \\&=\mathbb{E}[(a/n)^2\Var(\omega^\top A \omega)] + \Var(a/n \cdot \mathbb{E}[\omega^\top A \omega]) \\&= \frac{1}{n^2} \mathbb{E}[a^2] \cdot \Var(\omega^\top A \omega) + \frac{1}{n^2} \Var(a) |\mathbb{E} [\omega^\top A \omega]|^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3026a100436c6b4e822b5fd2468829ce_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[a^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0d927097d672c6d6e8d5be05ee727ce2_l3.png "Rendered by QuickLaTeX.com") and
and  are known quantities that
are known quantities that ![\[\mathbb{E}[a^2] = n(n+2), \quad \Var(a) = 2n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3fc9d94ca5542ae0cb9b24a30efbd46c_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E} [\omega^\top A \omega] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c72abe7ce9b67a916df0826948bef417_l3.png "Rendered by QuickLaTeX.com") . Plugging these all in, we get
. Plugging these all in, we get![\[2\left\|A\right\|_{\rm F}^2 = \frac{n+2}{n} \Var(\omega^\top A\omega) + \frac{2}{n} |\tr A|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-28c12df3dfc463c71106c87ab576a450_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\omega^\top A\omega) = \frac{2n}{n+2} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n}|\tr A|^2\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-65eb536bbaf2ab7b920da7cbdc6df709_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\omega^*A \omega) = \Var \left( \sum_{i=1}^n \lambda_i |\omega_i|^2 \right) = \sum_{i=1}^n \Var(|\omega_i|^2) \lambda_i^2 = \sum_{i=1}^n \lambda_i^2 = \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8373d673ee36f506deb78ccb637486a3_l3.png "Rendered by QuickLaTeX.com")
 is a
is a  ) reveals that
) reveals that![\[\Var\left( \omega^* A \omega \right) = \Var \left( \sum_{i<j} 2 \Re(A_{ij} \overline{\omega_i} \omega_j) \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-45fafcf974267732a375287d65a7cd4b_l3.png "Rendered by QuickLaTeX.com")
 are
are ![\[\Var\left( \omega^* A \omega \right) = \Var \left( \sum_{i<j} 2 \Re(A_{ij} \overline{\omega_i} \omega_j) \right) = 4\sum_{i<j} \Var \left( \Re(A_{ij} \overline{\omega_i} \omega_j) \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-51d809d4c6d2210932fcaf3b06b3f8b5_l3.png "Rendered by QuickLaTeX.com")
 is uniformly distributed on the complex unit circle, we can assume without loss of generality that
is uniformly distributed on the complex unit circle, we can assume without loss of generality that  . Thus, letting
. Thus, letting  be uniform on the complex unit circle,
be uniform on the complex unit circle,![\[\Var\left( \omega^* A \omega \right) = 4\sum_{i<j} \Var \left( |A_{ij}|\Re(\phi)) \right) = 4\Var\left( \Re(\phi) \right)\sum_{i<j}|A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b4a70df99004eec8eb6f05d26467c351_l3.png "Rendered by QuickLaTeX.com")
![\[1 = \Var(\phi) = \Var(\Re \phi) + \Var(\Im \phi) = 2 \Var(\Re \phi)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-014c1792fb787b53c5b17ba4c76af90e_l3.png "Rendered by QuickLaTeX.com")
 . Thus
. Thus![\[\Var\left( \omega^* A \omega \right) = 2 \sum_{i<j}|A_{ij}|^2 = \sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-07146eb21d91f21f747ea6a1bf092ea5_l3.png "Rendered by QuickLaTeX.com")
 and
and  denote the real and imaginary parts of a vector or matrix, taken entrywise. The key insight is that if
denote the real and imaginary parts of a vector or matrix, taken entrywise. The key insight is that if ![\[\mathscr{R}(\omega) := \twobyone{\mathfrak{R}(\omega)}{\mathfrak{I}(\omega)}\in\real^{2n} \quad \text{is a uniform random vector on the real sphere of radius $\sqrt{n}$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-13581c61bcdb79519cd801b56723d912_l3.png "Rendered by QuickLaTeX.com")
 .
. . To do this, recall that one way of representing complex numbers is by
. To do this, recall that one way of representing complex numbers is by  matrices:
matrices:![\[a + bi \iff \twobytwo{a}{-b}{b}{a}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06a101458da1c31ea0869cfa3dbd4b73_l3.png "Rendered by QuickLaTeX.com")
![\[\mathscr{R}(A) = \twobytwo{\mathfrak{R}(A)}{-\mathfrak{I}(A)}{\mathfrak{I}(A)}{\mathfrak{R}(A)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e4558ae7ba06d0d95db1ff3665f81093_l3.png "Rendered by QuickLaTeX.com")
![\[\mathscr{R}(A + B) = \mathscr{R}(A) + \mathscr{R}(B), \quad \mathscr{R}(A\cdot B) = \mathscr{R}(A) \cdot \mathscr{R}(B)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ca41602dcbf4ab0b1d2f1d2d19cb3156_l3.png "Rendered by QuickLaTeX.com")
 . A short calculation reveals
. A short calculation reveals![\[\omega^*A\omega = \mathscr{R}(\omega)^\top \mathscr{R}(A)\mathscr{R}(\omega) .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9cc7eb8d1c7b5726d44b062936cd4e59_l3.png "Rendered by QuickLaTeX.com")
 is a uniform random vector on the sphere of radius
is a uniform random vector on the sphere of radius  . Thus, by the variance formula for the real sphere, we get
. Thus, by the variance formula for the real sphere, we get![\[\Var(\omega^*A\omega) = \Var[(\sqrt{2}\mathscr{R}(\omega))^\top (\mathscr{R}(A)/2)(\sqrt{2}\mathscr{R}(\omega) )] = \frac{4n}{2n+2} \left[ \|\mathscr{R}(A)/2\|_{\rm F}^2 - \frac{1}{8n}(\tr\mathscr{R}(A))^2 \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc8feeeb958c69c6eb239478674f6748_l3.png "Rendered by QuickLaTeX.com")
 and
and  . Plugging this in, we obtain
. Plugging this in, we obtain![\[\Var(\omega^*A\omega)= \frac{n}{n+1} \left[ \|A\|_{\rm F}^2 - \frac{1}{n}(\tr A)^2 \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f48e0eebc5a1b52ff9357b83d348b1f6_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} [(\omega^*A \omega)^2].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c5a380c3abc691bdcc8542ebe3f48c0f_l3.png "Rendered by QuickLaTeX.com")
 . For those unfamiliar with the tensor product, the main properties we will be using
. For those unfamiliar with the tensor product, the main properties we will be using ![\[(A\otimes B) (C\otimes D) = (AB) \otimes (CD), \quad \tr(A\otimes B) = \tr A \cdot \tr B. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-175cabf924824dc07b5a92e57eab86c1_l3.png "Rendered by QuickLaTeX.com")
![\[\omega^* A \omega = \tr (\omega^*A\omega) = \tr (A \omega\omega^*).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ce640b29ebfc673e441b2339b9ec28ec_l3.png "Rendered by QuickLaTeX.com")
![\[(\omega^*A\omega)^2 = (\tr [A \omega\omega^*])^2 = \tr [A\omega\omega^* \otimes A\omega\omega^*] = \tr [(A\otimes A) (\omega\omega^* \otimes \omega\omega^*)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-be12748493beb02472a022c5efc1f855_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[(\omega^*A\omega)^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-77022381e46a04de0881046ee13a39a6_l3.png "Rendered by QuickLaTeX.com") , it will be sufficient to evaluate
, it will be sufficient to evaluate ![\mathbb{E}[\omega\omega^* \otimes \omega\omega^*]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3bddca06f7eee0b7a100634abdcf084f_l3.png "Rendered by QuickLaTeX.com") . Forunately, there is a useful formula for these expectation provided by a field of mathematics known as representation theory (see Lemma 1 in
. Forunately, there is a useful formula for these expectation provided by a field of mathematics known as representation theory (see Lemma 1 in ![\[\mathbb{E}[ \omega\omega^* \otimes \omega\omega^*] = \frac{2n}{n+1} \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5c39d88134e7a3628744cf2509294902_l3.png "Rendered by QuickLaTeX.com")
 is the
is the  . Therefore, we have that
. Therefore, we have that![\[\mathbb{E}[(\omega^*A\omega)^2] = \tr [(A\otimes A) \mathbb{E}(\omega\omega^* \otimes \omega\omega^*)] = \frac{2n}{n+1} \tr [(A\otimes A) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-42f3b90989c6405e98ff6d79a7fc7906_l3.png "Rendered by QuickLaTeX.com")
![\[\tr \left[(A\otimes B) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}\right] = \frac{1}{2} \left( \tr(AB) + \tr A \cdot \tr B \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-09896bb79140d5decdac0b302f6d761c_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\Var(\omega^* A \omega) &= \mathbb{E}[(\omega^*A\omega)^2] - (\mathbb{E}[\omega^*A\omega])^2 \\&= \frac{2n}{n+1}\tr [(A\otimes A) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}] - (\tr A)^2 \\&= \frac{n}{n+1}\left[ \tr A^2 + (\tr A)^2 \right] - (\tr A)^2 \\&= \frac{n}{n+1}\left[ \left\|A\right\|_{\rm F}^2 - \frac{1}{n} (\tr A)^2 \right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6ab261cb7e9ad3e0f3a218fd75ad8ebf_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E}[\omega\omega^\top] = I\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-69502d5f0214a4d4cc39484a9725fe8c_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[\omega_i\omega_j] = \delta_{ij}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-72403dc927db310a6df481cb0eea9a11_l3.png "Rendered by QuickLaTeX.com") , where
, where ![\begin{align*}\mathbb{E}[ (\omega^*A \omega)^2] &= \mathbb{E}\left[ \left( \sum_{i=1}^n A_{ii} \omega_i^2 +2 \sum_{i<j} A_{ij}\omega_i\omega_j) \right)^2\right] \\&= \sum_{i=1}^n A_{ii}^2 \mathbb{E}[\omega_i^4] + \sum_{i<j} (2A_{ii}A_{jj}+4A_{ij}^2) \mathbb{E}[\omega_i^2]\mathbb{E}[\omega_j^2] \\&= \sum_{i=1}^n A_{ii}^2 \mathbb{E}[\omega_i^4] + \sum_{i<j} (2A_{ii}A_{jj}+4A_{ij}^2) .\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2123a841fb0b78ce9edbbc2f4a198beb_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\omega^*A\omega) = \mathbb{E}[ (\omega^*A \omega)^2] - (\mathbb{E}[\omega^* A \omega])^2 = \sum_{i=1}^n A_{ii}^2 (\mathbb{E}[|\omega_i|^4]-1) + 4\sum_{i<j} A_{ij}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-95fe70ef6f036d673badde1304da735e_l3.png "Rendered by QuickLaTeX.com")
 as small as possible. Since
as small as possible. Since  , the smallest possible value for
, the smallest possible value for  , which is obtained by populating
, which is obtained by populating ![\[\omega = a \cdot s \quad \text{for} \quad a \in [0,+\infty), \: s \in\{x\in \field^n : x^*x = n \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9378439b80ef13bf2dff81e656ed01b3_l3.png "Rendered by QuickLaTeX.com")
 with a vector
with a vector ![\[\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega}) \le \sup_{A\in\mathscr{A}} \Var(\omega^*A\omega \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-110fc8f9b36ec9b032825e0b3c6e62ca_l3.png "Rendered by QuickLaTeX.com")
![\[\tilde{\omega} = a\cdot t \quad \text{and}\quad t\sim \text{Uniform} \{ x \in \field^n :x^*x = n \} \quad \text{is independent of $a$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-af90470a85bcf4c3d1c303669a811101_l3.png "Rendered by QuickLaTeX.com")
 for a uniformly random
for a uniformly random ![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[(\tilde{\omega}^*A\tilde{\omega})^2] - (\tr A)^2\right]\\&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dd38727e24f3ef4e3ef1c84ae784acdd_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right] \\&\le \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0c4a1612eebf87861ced8ae83b61f92a_l3.png "Rendered by QuickLaTeX.com")
 for
for  is the same as the supremum of
is the same as the supremum of ![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&\le \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right] \\&= \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*As] - (\tr A)^2\right] \\&= \sup_{A\in\mathscr{A}} \Var(\omega^*A\omega).\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49501bf09d0be7059918505c0f975c21_l3.png "Rendered by QuickLaTeX.com")
![\[\sup_{A\in\mathscr{A}} \Var(t^*At) \le \sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega}), \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2c67d927a461f04c98b5f1b76e7bda02_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\Var(\tilde{\omega}^*A\tilde{\omega})&= \Var(a^2\cdot t^*At) \\&= \mathbb{E}[\Var(a^2\cdot t^* A t \mid a)] + \Var(\mathbb{E}[a^2\cdot t^* A t \mid a]) \\&= \mathbb{E}[a^4]\Var(t^* A t )+ (\tr A)^2\Var(a^2) \\&\ge \mathbb{E}[a^4]\Var(t^* A t ).\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff41cdacd7b61186030f66166abeeff2_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[t^*At] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d160f311b7c70e20eb531b28c90cd135_l3.png "Rendered by QuickLaTeX.com") . By definition,
. By definition, ![\[\mathbb{E}[a^2] = \frac{1}{n}\mathbb{E}[\omega^*\omega] = \frac{1}{n} \mathbb{E}[\tr (\omega\omega^*)] = \frac{1}{n} \tr (\mathbb{E}[\omega\omega^*]) = \frac{\tr I}{n} = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fed236a34834a474032da2b77177dac9_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E}[a^4] = \mathbb{E}[(a^2)^2] \ge (\mathbb{E}[a^2])^2 = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-63438e2a64c576b7502e66c1f165a3bb_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\tilde{\omega}^*A\tilde{\omega}) \ge \mathbb{E}[a^4]\Var(t^* A t ) \ge \Var(t^*At) \quad \text{for every }A,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff80c63bbb1cac110454cdea808ca304_l3.png "Rendered by QuickLaTeX.com")
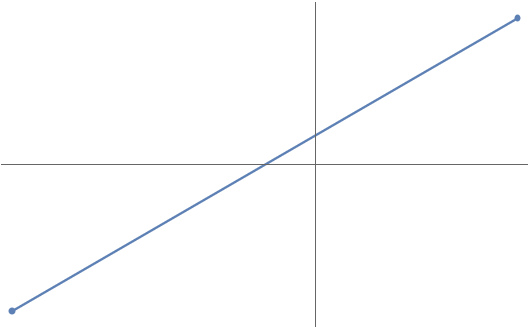
 :
: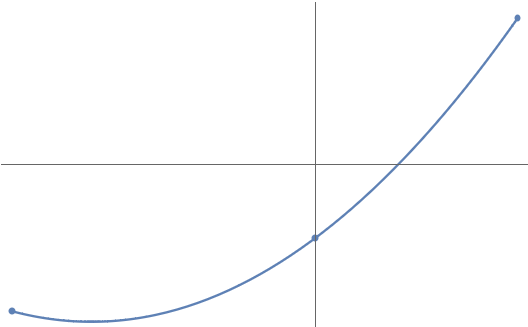
 points
points , and
, and  .
. :
: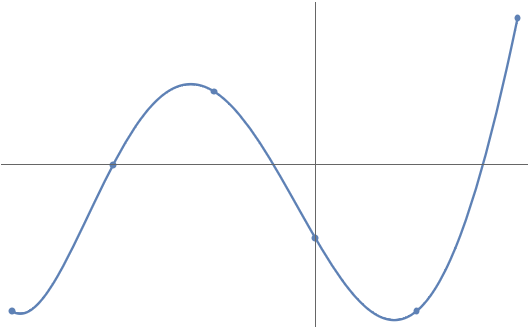
 on the interval
on the interval ![[-\pi,\pi]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-441294250886cf4d074975f1f08d0ed1_l3.png "Rendered by QuickLaTeX.com") to visually near-perfect accuracy by connecting the dots between seven points
to visually near-perfect accuracy by connecting the dots between seven points  :
: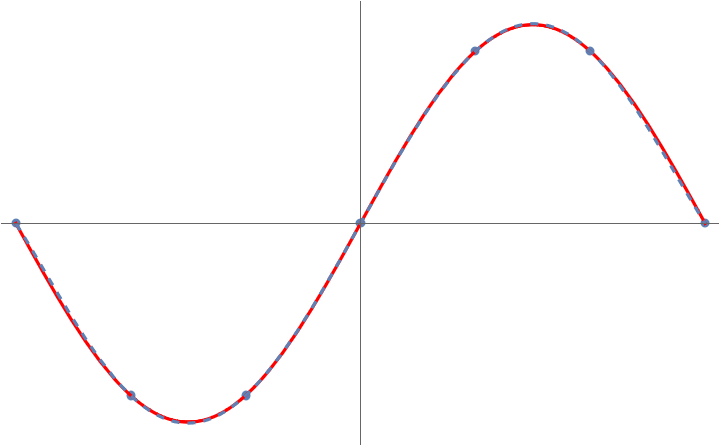
 on the interval
on the interval ![[-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7e5005e1a5c93f262ad8df6aaa18b0bc_l3.png "Rendered by QuickLaTeX.com") :
: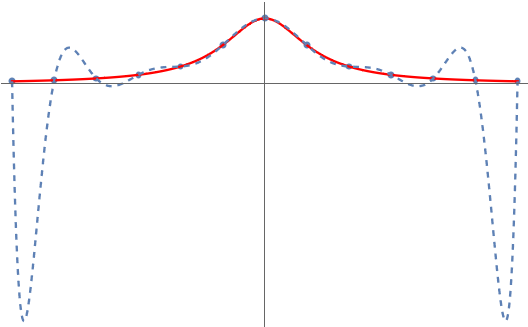
 is terrible! What’s going on? Why doesn’t polynomial interpolation work here? Can we fix it? The answer to the last question is yes and the solution is Chebyshev polynomials.
is terrible! What’s going on? Why doesn’t polynomial interpolation work here? Can we fix it? The answer to the last question is yes and the solution is Chebyshev polynomials.

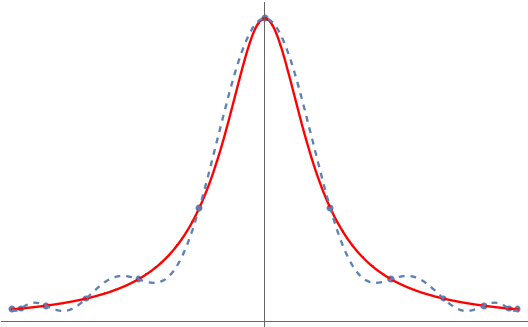
 , like this for ourselves. We’ll assume the interpolation points are given by a function
, like this for ourselves. We’ll assume the interpolation points are given by a function  so we can form the polynomial interpolant for any desired polynomial degree
so we can form the polynomial interpolant for any desired polynomial degree  being \textit{injective} so that the points
being \textit{injective} so that the points  are guaranteed to be distinct.
are guaranteed to be distinct. , the points look like this:
, the points look like this: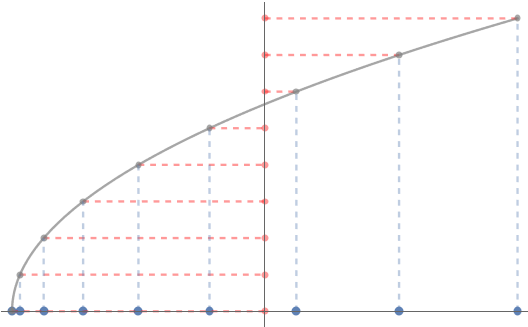

 (shown on horizontal axis)
(shown on horizontal axis) .
.
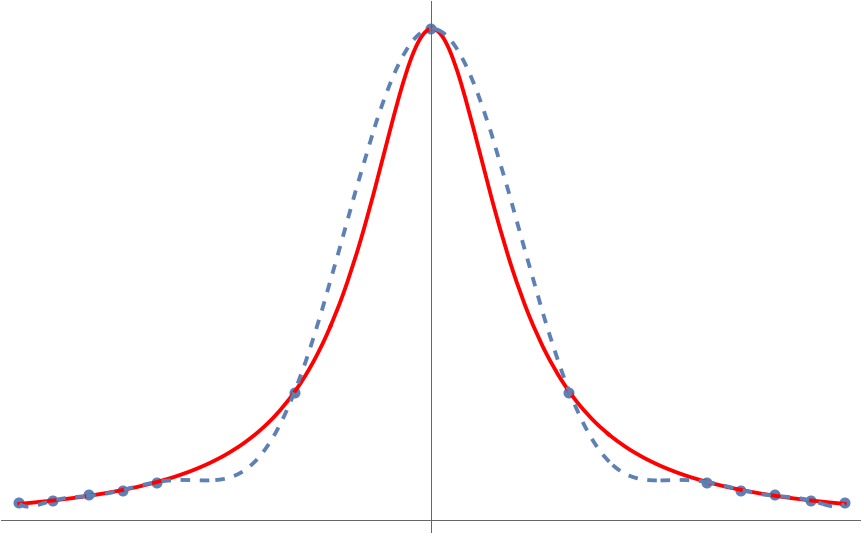
 , we see that these interpolation points now lead to a failure to approximate the function at the middle of the interval, even if we use a lot of interpolation points!
, we see that these interpolation points now lead to a failure to approximate the function at the middle of the interval, even if we use a lot of interpolation points!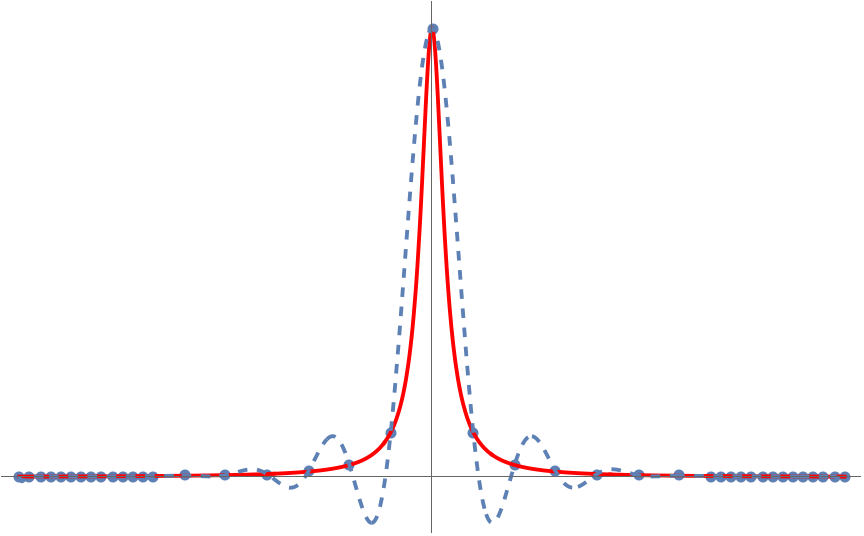
 and
and  are, the more clustered the points will be. For instance,
are, the more clustered the points will be. For instance,


 , and
, and ,
,  .
. . This function yields points which are more clustered at the endpoints:
. This function yields points which are more clustered at the endpoints:


 which not too rough (i.e.,
which not too rough (i.e.,  .
. gives the
gives the  radians along the unit circle. This means that the Chebyshev points are the
radians along the unit circle. This means that the Chebyshev points are the  ).
).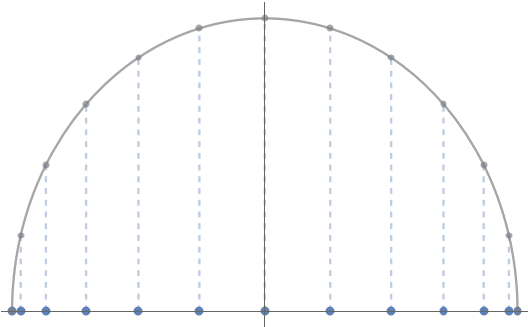
 look like as a function of the angle
look like as a function of the angle  to
to 
 , is a polynomial in the variable
, is a polynomial in the variable 
 always take the form
always take the form

 units of frequency
units of frequency  units of frequency
units of frequency  units of frequency
units of frequency  with exponentially small errors in the number of interpolation points used. By contrast, functions with kinks like
with exponentially small errors in the number of interpolation points used. By contrast, functions with kinks like  are approximated with errors which decay much more slowly. See
are approximated with errors which decay much more slowly. See 
 is
is  since multiple
since multiple  , between which the cosine function is
, between which the cosine function is 
 , we’ve instead written
, we’ve instead written 
 are known as the
are known as the  are known as the Chebyshev polynomials
are known as the Chebyshev polynomials 

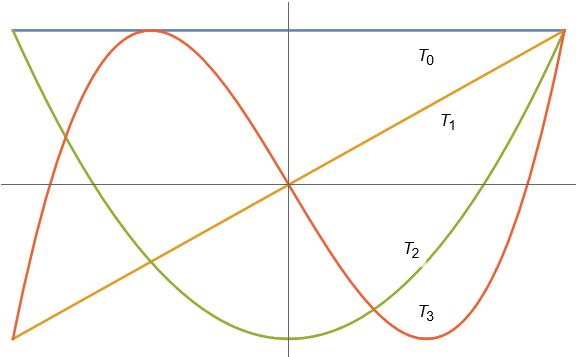
 for
for 
 and
and  are both polynomials, every Chebyshev polynomial is as well.
are both polynomials, every Chebyshev polynomial is as well.![\[p^\circ(\theta) = b_n\cos(n\theta) + \cdots + b_1\cos\theta + b_0\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5ec96f888a19309f94dee95bc8234515_l3.png "Rendered by QuickLaTeX.com")
![\[p(x) = b_nT_n(x) + \cdots + b_1 T_1(x) + b_0.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bb9fcd901283bb933882efd81c4c5e26_l3.png "Rendered by QuickLaTeX.com")
 are orthogonal to each other; are the Chebyshev polynomials?
are orthogonal to each other; are the Chebyshev polynomials? matrix
matrix  is as close to
is as close to  . That is, we seek to
. That is, we seek to 
 of
of  of
of  , our model predicts the output
, our model predicts the output  is approximately
is approximately  . The vector
. The vector  is as small as it could possibly be for all choices of coefficient vectors
is as small as it could possibly be for all choices of coefficient vectors 
 is invertible and the unique least squares solution to (1) is given by
is invertible and the unique least squares solution to (1) is given by  . We assume that
. We assume that  and solve (2) using a linear solver like
and solve (2) using a linear solver like  on a computer using a well-designed piece of software, one obtains an approximate solution
on a computer using a well-designed piece of software, one obtains an approximate solution  which is, after accounting for the accumulation of rounding errors, close to
which is, after accounting for the accumulation of rounding errors, close to  is. The “niceness” of a matrix
is. The “niceness” of a matrix  .
. of the largest and smallest
of the largest and smallest 
 corresponds to the fact we have roughly 16 decimal digits of accuracy in double precision floating point arithmetic. Thus, if the condition number of
corresponds to the fact we have roughly 16 decimal digits of accuracy in double precision floating point arithmetic. Thus, if the condition number of  , then we should expect around
, then we should expect around  digits of accuracy in our computed solution.
digits of accuracy in our computed solution. . We would hope that we can solve the least squares problem with an accuracy like the rule-of-thumb error bound (3) we had for linear systems of equations, namely a bound like
. We would hope that we can solve the least squares problem with an accuracy like the rule-of-thumb error bound (3) we had for linear systems of equations, namely a bound like  . But this is not the kind of accuracy we get for the least squares problem when we solve it using the normal equations. Instead, we get accuracy like
. But this is not the kind of accuracy we get for the least squares problem when we solve it using the normal equations. Instead, we get accuracy like
 , then the normal equations give us absolute nonsense for
, then the normal equations give us absolute nonsense for  times larger than
times larger than  . So even if we solve the least squares problem with QR factorization, we still get a squared condition number in our error bound, but this condition number squared is multiplied by the residual
. So even if we solve the least squares problem with QR factorization, we still get a squared condition number in our error bound, but this condition number squared is multiplied by the residual  , which is small if the least squares fit is good. The least squares solution is usually only interesting when the residual is small, thus justifying dropping it in the rule of thumb.
, which is small if the least squares fit is good. The least squares solution is usually only interesting when the residual is small, thus justifying dropping it in the rule of thumb.
 , etc.
, etc. denote the temperature of the material at point
denote the temperature of the material at point  . Laplace’s equation states that, at any point
. Laplace’s equation states that, at any point 

 .
. on the boundary to produce the electric potential
on the boundary to produce the electric potential  be the region of interest and
be the region of interest and  its boundary. Denote points
its boundary. Denote points  , with the length of
, with the length of  denoted
denoted  . The double layer potential satisfies
. The double layer potential satisfies
 denotes the directional derivative taken in the direction normal (perpendicular) to the surface at the point
denotes the directional derivative taken in the direction normal (perpendicular) to the surface at the point  . Note we choose a unit system for
. Note we choose a unit system for  at points
at points 
 centered at
centered at 


 square matrix and
square matrix and  ,
,  in memory and multiplying it with
in memory and multiplying it with  .
. .
. . Specifically, our first equation can be rewritten as
. Specifically, our first equation can be rewritten as




 , we have that
, we have that

 from
from  but, as we did last section, let’s first consider the reverse. To compute
but, as we did last section, let’s first consider the reverse. To compute 


 times equation (8) to equation (7). Following last section, we now instead eliminate
times equation (8) to equation (7). Following last section, we now instead eliminate  from equation (8) using equation (7). To do this, we need to integrate equation (7) in order to cancel the integral in equation (8):
from equation (8) using equation (7). To do this, we need to integrate equation (7) in order to cancel the integral in equation (8):
 times this integrated equation to equation (8) yields
times this integrated equation to equation (8) yields


 using the
using the 
 for the
for the  for the normal vector to
for the normal vector to  for the
for the  for the
for the  is a so-called
is a so-called  . Therefore, (9) simplifies to
. Therefore, (9) simplifies to

 for points
for points 
 is some region in space,
is some region in space,  ,
,  is a nonzero constant. Such an integral equation is said to be of the
is a nonzero constant. Such an integral equation is said to be of the  ,
,  ,
,  , and
, and  . We say the kernel
. We say the kernel  if
if 
 .
. and
and  .
. ):
):
 denote the identity operator on functions: It takes as inputs function
denote the identity operator on functions: It takes as inputs function  denote the “integration against
denote the “integration against  operator”: It takes as input a function
operator”: It takes as input a function  .
.


 system of linear equations. One can use this as a numerical method to solve second-kind integral equations: First, we
system of linear equations. One can use this as a numerical method to solve second-kind integral equations: First, we  by a separable kernel of a modest rank
by a separable kernel of a modest rank  -decompositions, sparse component analysis, and the blind separation of sums of exponentials
-decompositions, sparse component analysis, and the blind separation of sums of exponentials![\[f(t) = a_0 \cos(\omega_0 t - \phi_0) + a_1 \cos(\omega_1 t - \phi_1) + a_2 \cos(\omega_2 t - \phi_2). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8e44bb98be117230aa4cd5148d841129_l3.png "Rendered by QuickLaTeX.com")
 ,
,  , and
, and  .
. which we assume are equally spaced
which we assume are equally spaced![\[t_j = j\Delta t \quad \textnormal{for} \quad j = 0,1,2,\ldots,n-1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-814820bd1ef46e2fee855eb68600d927_l3.png "Rendered by QuickLaTeX.com")
![\[f_j = f(t_j) \quad \textnormal{for} \quad j = 0,1,2,\ldots,n-1\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a0b9511cab842078a47a27836c5821e1_l3.png "Rendered by QuickLaTeX.com")
 , where
, where  it will be helpful to rewrite our formula (1) for
it will be helpful to rewrite our formula (1) for ![\[\cos \alpha = \frac{\mathrm{e}^{\mathrm{i} \alpha}+\mathrm{e}^{-\mathrm{i} \alpha}}{2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ffb0475e120f3b8c6340c432631c5962_l3.png "Rendered by QuickLaTeX.com")
![\[a_0 \cos(\omega_0 t - \phi_0) = d_0 \mathrm{e}^{\mathrm{i} \omega_0t} + d_1 \mathrm{e}^{-\mathrm{i} \omega_0t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1683f7679dca96f66c9221332d5e0bdb_l3.png "Rendered by QuickLaTeX.com")
 and
and  are complex coefficients
are complex coefficients  and
and  which contain the same information as the original parameters
which contain the same information as the original parameters  and
and  . Now notice that we are only interest in values
. Now notice that we are only interest in values  which are multiples of the spacing
which are multiples of the spacing  . Thus, our frequency component can be further rewritten as
. Thus, our frequency component can be further rewritten as![\[a_0 \cos(\omega_0 t_j - \phi_0) = d_0 z_0^j + d_1 z_1^j\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-269ef790089c772d8c424bd62aa1707e_l3.png "Rendered by QuickLaTeX.com")
 and
and  . Performing these reductions, our samples
. Performing these reductions, our samples  take the form
take the form![\[f_j = d_0 z_0^j + d_1 z_1^j + \cdots + d_5 z_5^j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a32d21e3f31302be06340549f7bf65b2_l3.png "Rendered by QuickLaTeX.com")
 and
and  in the relation (2) from a small number of measurements
in the relation (2) from a small number of measurements  . This can reveal patterns in the sequence which are less obvious when it is represented as given just as numbers. For instance, any seven columns of
. This can reveal patterns in the sequence which are less obvious when it is represented as given just as numbers. For instance, any seven columns of  which can be much larger than seven. In addition, as we will soon effectively exploit later, vectors in the nullspace of
which can be much larger than seven. In addition, as we will soon effectively exploit later, vectors in the nullspace of ![\[H = \begin{bmatrix} f_0 & f_1 & f_2 & \cdots & f_{(n-1)/2} \\f_1 & f_2 & f_3 & \cdots & f_{(n-1)/2+1} \\f_2 & f_3 & f_4 & \cdots & f_{(n-1)/2+2} \\\vdots & \vdots & \vdots & \ddots & \vdots \\f_{(n-1)/2} & f{(n-1)/2+1} & f{(n-1)/2+2} & \cdots & f_{n-1} \end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-42f02441b9724e66513f1e422c797113_l3.png "Rendered by QuickLaTeX.com")
 . Observe that the entry in position
. Observe that the entry in position  of
of  ,
,  . (We use a
. (We use a  using (2):
using (2):![\[H_{ij} = f_{i+j} = \sum_{k=0}^5 d_k z_k^{i+j} = \sum_{k=0}^5 d_k z_k^i \cdot z_k^j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c40e19f060d1924406303ffb887b2153_l3.png "Rendered by QuickLaTeX.com")
 was just begging to be factorized as
was just begging to be factorized as  , which we did. Equation (4) almost looks like the formula for the product of two matrices with entries
, which we did. Equation (4) almost looks like the formula for the product of two matrices with entries  , so it makes sense to introduce the
, so it makes sense to introduce the  matrix
matrix  with entry
with entry  . This is a so-called
. This is a so-called ![\[V = \begin{bmatrix}z_0^0 & z_0^1 & z_0^2 & \cdots & z_0^{(n-1)/2} \\z_1^0 & z_1^1 & z_1^2 & \cdots & z_1^{(n-1)/2} \\\vdots & \vdots & \vdots & \ddots & \vdots \\z_5^0 & z_5^1 & z_5^2 & \cdots & z_5^{(n-1)/2}\end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a1d818b7fab7cb659d2a20c9f94bc743_l3.png "Rendered by QuickLaTeX.com")
 diagonal matrix
diagonal matrix  , the formula (4) for
, the formula (4) for  , the factor
, the factor ![\[H = V^\top D V. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2a7b807a19d51a8bd8d8de2e04aac3f3_l3.png "Rendered by QuickLaTeX.com")
 describing our sampled recording
describing our sampled recording  , determining all the frequencies would be very straightforward.
, determining all the frequencies would be very straightforward. of
of ![\[f_m = c_{k-1} f_{m-1} + \cdots + c_1f_{m-k+1}+ c_0 f_{m-k} \quad \textnormal{for every} \quad m = k,\,{k+1},\,{k+2},\cdots. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-62d879500458ed1476a089397147f5c4_l3.png "Rendered by QuickLaTeX.com")
 because there are six terms in the formula (2) for
because there are six terms in the formula (2) for  , such a recurrence will allow us to determine the parameters
, such a recurrence will allow us to determine the parameters  . A very good rule of thumb in applied mathematics is to always write down linear equations in matrix–vector notation in see how it looks. Doing this, we obtain
. A very good rule of thumb in applied mathematics is to always write down linear equations in matrix–vector notation in see how it looks. Doing this, we obtain![\[\begin{bmatrix} f_6 \\ f_7 \\ \vdots \\ f_{n-1} \end{bmatrix} = \underbrace{\begin{bmatrix} f_0 & f_1 & f_2 & f_3 & f_4 & f_5 \\ f_1 & f_2 & f_3 & f_4 & f_5 & f_6 \\ \vdots & \vdots & \vdots & \vdots & \vdots & \vdots \\ f_{n-7} & f_{n-6} & f_{n-5} & f_{n-4} & f_{n-3} & f_{n-2} \end{bmatrix}}_{=F}\begin{bmatrix} c_0 \\ c_1 \\ \vdots \\ c_5 \end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-aa5891d632cb53b7b006e08328f5d3a0_l3.png "Rendered by QuickLaTeX.com")
 . Unlike
. Unlike  going forward.
going forward. would also be fine for our purposes, but we assume
would also be fine for our purposes, but we assume ![\[f_{6 \, :\, n-1} = F c, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-206cb0e274493646eb686f2fc6b773df_l3.png "Rendered by QuickLaTeX.com")
 for the vector on the left-hand side of (7) and collected the recurrence coefficients
for the vector on the left-hand side of (7) and collected the recurrence coefficients  ), the system equations (8) has more equations than unknowns. Fortunately, we are not in the typical case. Despite the fact that we have more equations than unknowns, the linear equations (8) have a unique solution
), the system equations (8) has more equations than unknowns. Fortunately, we are not in the typical case. Despite the fact that we have more equations than unknowns, the linear equations (8) have a unique solution  In particular, the matrix on the right-hand side of this equation is guaranteed to be nonsingular under our assumptions. Using the Vandermonde decomposition, can you see why?
In particular, the matrix on the right-hand side of this equation is guaranteed to be nonsingular under our assumptions. Using the Vandermonde decomposition, can you see why? ; we seek to find all complex numbers
; we seek to find all complex numbers  gives
gives![\[z^6 = c_0 z^5 + c_1 z^4 + \cdots + c_6. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c36ec055ae832307db7d77cb25d469a_l3.png "Rendered by QuickLaTeX.com")
![\[\begin{bmatrix}f_0 \\ f_1 \\ \vdots \\ f_{n-1}\end{bmatrix} = \begin{bmatrix}1 & 1 & \cdots & 1 \\ z_0 & z_1 & \cdots & z_5 \\ \vdots & \vdots & \ddots & \vdots \\ z_0^{n-1} & z_1^{n-1} & \cdots & z_5^{n-1}\end{bmatrix} \begin{bmatrix}d_0 \\ d_1 \\ \vdots \\ d_{n-1}.\end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b1cdea36621ca6c0c1c791764d993fea_l3.png "Rendered by QuickLaTeX.com")
 . So far, the benefits of the matrix factorization perspective have yet to really reveal themselves.
. So far, the benefits of the matrix factorization perspective have yet to really reveal themselves. needed to describe the samples and the representation of the samples as a mixture of exponentials is uniquely determined only if the matrix
needed to describe the samples and the representation of the samples as a mixture of exponentials is uniquely determined only if the matrix  ,
,  , and
, and  . (Here, the superscript denotes an index rather than differentiation.) Each signal is a weighted mixture of three sources
. (Here, the superscript denotes an index rather than differentiation.) Each signal is a weighted mixture of three sources  ,
,  , and
, and  , each of which plays a musical chord of three notes (thus representable as a sum of cosines as in (1)). One can think of the sources of being produced three different musical instruments at different places in a room and the recordings
, each of which plays a musical chord of three notes (thus representable as a sum of cosines as in (1)). One can think of the sources of being produced three different musical instruments at different places in a room and the recordings  for each recording
for each recording  and form each collection of samples into a Hankel matrix
and form each collection of samples into a Hankel matrix![\[H^{(\ell)} = \begin{bmatrix} f_0^{(\ell)} & f_1^{(\ell)} & f_2^{(\ell)} & \cdots & f_{(n-1)/2}^{(\ell)} \\f_1^{(\ell)} & f_2^{(\ell)} & f_3^{(\ell)} & \cdots & f_{(n-1)/2+1}^{(\ell)} \\f_2^{(\ell)} & f_3^{(\ell)} & f_4^{(\ell)} & \cdots & f_{(n-1)/2+2}^{(\ell)} \\\vdots & \vdots & \vdots & \ddots & \vdots \\f_{(n-1)/2}^{(\ell)} & f_{(n-1)/2+1}^{(\ell)} & f_{(n-1)/2+2}^{(\ell)} & \cdots & f_{n-1}^{(\ell)} \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5c3bb6afd39df0eba56eaeee8c08a77f_l3.png "Rendered by QuickLaTeX.com")
 ,
,  , and
, and  as slices of a
as slices of a  . A tensor,
. A tensor,  array given by
array given by![\[\mathcal{H}(:,:,\ell) = H^{(\ell)} \quad \textnormal{for} \quad \ell=1,2,3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7a46aa5e41df12034dc3c63905a626ba_l3.png "Rendered by QuickLaTeX.com")
![\[H = V^\top DV = \tilde{V}^\top \tilde{D} \tilde{V}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a480b33a17f5c4bbe9f3e057a9350924_l3.png "Rendered by QuickLaTeX.com")
 can be uniquely determined from the measurements
can be uniquely determined from the measurements  rows,
rows,  ‘s are all uniquely determined.
‘s are all uniquely determined.  and perhaps
and perhaps  ,
,  , etc.
, etc. This matrix has rank-two but no rank-two confluent Vandermonde decomposition. The issue is that when extended to an infinite Hankel matrix
This matrix has rank-two but no rank-two confluent Vandermonde decomposition. The issue is that when extended to an infinite Hankel matrix  this (infinite!) matrix has a rank exceeding the size of the original Hankel matrix
this (infinite!) matrix has a rank exceeding the size of the original Hankel matrix  into a Hankel matrix, it can also be useful to form them into a
into a Hankel matrix, it can also be useful to form them into a ![\[T = \begin{bmatrix} f_{(n-1)/2} & f_{(n-1)/2+1} & f_{(n-1)/2+2} & \cdots & f_{n-1} \\\\f_{(n-1)/2-1} & f_{(n-1)/2} & f_{(n-1)/2+1} & \cdots & f_{n-2} \\\\f_{(n-1)/2-2} & f_{(n-1)/2-1} & f_{(n-1)/2} & \cdots & f_{n-3} \\\\\vdots & \vdots & \vdots & \ddots & \vdots \\\\ f_0 & f_1 & f_2 & \cdots & f_{(n-1)/2} \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a99cb184de9f3b17cda900a0997b483c_l3.png "Rendered by QuickLaTeX.com")
 has the appealing propery that the matrix–vector product
has the appealing propery that the matrix–vector product  computes a
computes a ![\[T = \operatorname{ReversedRows}(V^\top) \cdot DV.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-66be95d184f371b43d13547ce6eb5a81_l3.png "Rendered by QuickLaTeX.com")
![\[T = V^* D V,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ef493ca7d7b0327e179da261e9f32ddb_l3.png "Rendered by QuickLaTeX.com")
 which are
which are  where
where  .
. such that
such that  .
. , which is a special case of the generalized eigenvalue problem with
, which is a special case of the generalized eigenvalue problem with  .
. denotes
denotes  are given by the
are given by the  . If we choose
. If we choose  is a (static) equilibrium configuration for which
is a (static) equilibrium configuration for which  whenever
whenever  .
.

 , where
, where  .
. can be written (uniquely) as linear combinations of solutions of the form
can be written (uniquely) as linear combinations of solutions of the form  . For several reasons, this is a less-than-appealing way of reducing a generalized eigenvalue problem to a standard eigenvalue problem. A better way, appropriate when
. For several reasons, this is a less-than-appealing way of reducing a generalized eigenvalue problem to a standard eigenvalue problem. A better way, appropriate when  , which also possesses the same eigenvalues as
, which also possesses the same eigenvalues as  .
. compare to those of
compare to those of  and
and  , where
, where  is the
is the  .) For the remainder of this post, let
.) For the remainder of this post, let  and
and  denote the perturbations.
denote the perturbations. , then
, then  . That is,
. That is,  is an eigenvalue of the pair
is an eigenvalue of the pair  . Lack of symmetry is an ugly feature in a mathematical theory, so we seek to smooth it out. After thinking a little bit, notice that we can phrase the generalized eigenvalue condition symmetrically as
. Lack of symmetry is an ugly feature in a mathematical theory, so we seek to smooth it out. After thinking a little bit, notice that we can phrase the generalized eigenvalue condition symmetrically as  with the associated eigenvalue being given by
with the associated eigenvalue being given by  . This observation may seem trivial at first, but let us collect it for good measure.
. This observation may seem trivial at first, but let us collect it for good measure. .
. ? The case
? The case  , this expression still makes sense: we’ve found a vector
, this expression still makes sense: we’ve found a vector  but
but  . It makes sense to consider
. It makes sense to consider  ! Dividing by zero should justifiably make one squeemish, but it really is quite natural in the case to treat
! Dividing by zero should justifiably make one squeemish, but it really is quite natural in the case to treat  .
. . Then, any
. Then, any  can reasonably considered an eigenvalue of
can reasonably considered an eigenvalue of  . In such a case, all complex numbers are simultaneously eigenvalues of
. In such a case, all complex numbers are simultaneously eigenvalues of  is identically zero for all
is identically zero for all  .
. . Thus, it’s better not to think of
. Thus, it’s better not to think of  for
for  . Geometrically, the set of all positive scalings of a point in two-dimensional space is precisely just a ray emanating from the origin.
. Geometrically, the set of all positive scalings of a point in two-dimensional space is precisely just a ray emanating from the origin.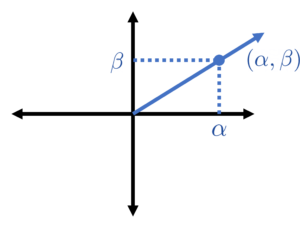
 and rearranging gives
and rearranging gives  —the eigenvalue
—the eigenvalue  , which is so important it is given a name: the
, which is so important it is given a name: the 
 and
and  . And by different scalings of the eigenvector
. And by different scalings of the eigenvector  and
and  by any positive factor we want. (This retroactively shows why it makes sense to only consider positive scalings of
by any positive factor we want. (This retroactively shows why it makes sense to only consider positive scalings of  . Thus, even though
. Thus, even though  lead to equivalent eigenvalues since
lead to equivalent eigenvalues since  ,
,  , this does not mean there exists
, this does not mean there exists  such that
such that  . Therefore, we only consider
. Therefore, we only consider  is so important that we give it a name: the
is so important that we give it a name: the  .
. is real, so we can pick a scaling in which both
is real, so we can pick a scaling in which both 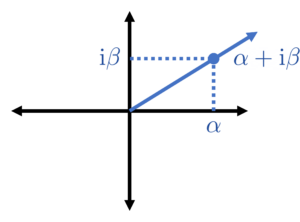
 is best thought of as a pair
is best thought of as a pair 
 .If we just went straight from one to the other, this reduction would appear like some crazy stroke of inspiration: why would I ever think to write down
.If we just went straight from one to the other, this reduction would appear like some crazy stroke of inspiration: why would I ever think to write down  , we have that
, we have that  : consequently,
: consequently,  is a small perturbation of
is a small perturbation of  is
is  as well and thus
as well and thus  . It is thus quite natural to assume the following definiteness condition:
. It is thus quite natural to assume the following definiteness condition: for all complex nonzero vectors
for all complex nonzero vectors  .
.  . However,
. However,  is not identically zero..
is not identically zero.. for all vectors
for all vectors  is just scaled by a positive factor by scaling the vector
is just scaled by a positive factor by scaling the vector  of a pair
of a pair  over all complex unit vectors
over all complex unit vectors  .
.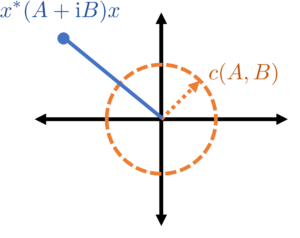
 ), the set of complex numbers
), the set of complex numbers  for all unit vectors
for all unit vectors  .
. to zero.
to zero. for a definite matrix pair
for a definite matrix pair  .
. for unit vectors
for unit vectors 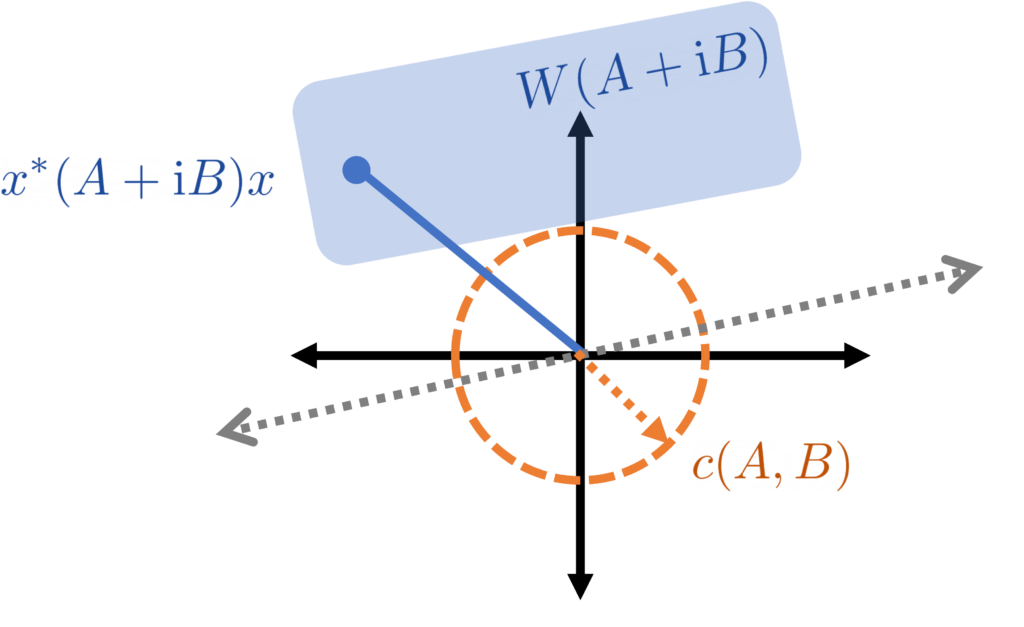
 radians). As such, to associate each ray a unique angle we need to pin down this indeterminacy in some fixed way. Moreover, this indeterminacy should play nice with the field of values
radians). As such, to associate each ray a unique angle we need to pin down this indeterminacy in some fixed way. Moreover, this indeterminacy should play nice with the field of values  of the perturbation. But in the last section, we say that each of these field of angles lies (strictly) on one half of the complex plane. Thus, we can find a ray
of the perturbation. But in the last section, we say that each of these field of angles lies (strictly) on one half of the complex plane. Thus, we can find a ray  for some real number
for some real number  . The argument is multi-valued since
. The argument is multi-valued since  is an argument for
is an argument for  as long as
as long as  . If
. If  an eigenangle.
an eigenangle. .
.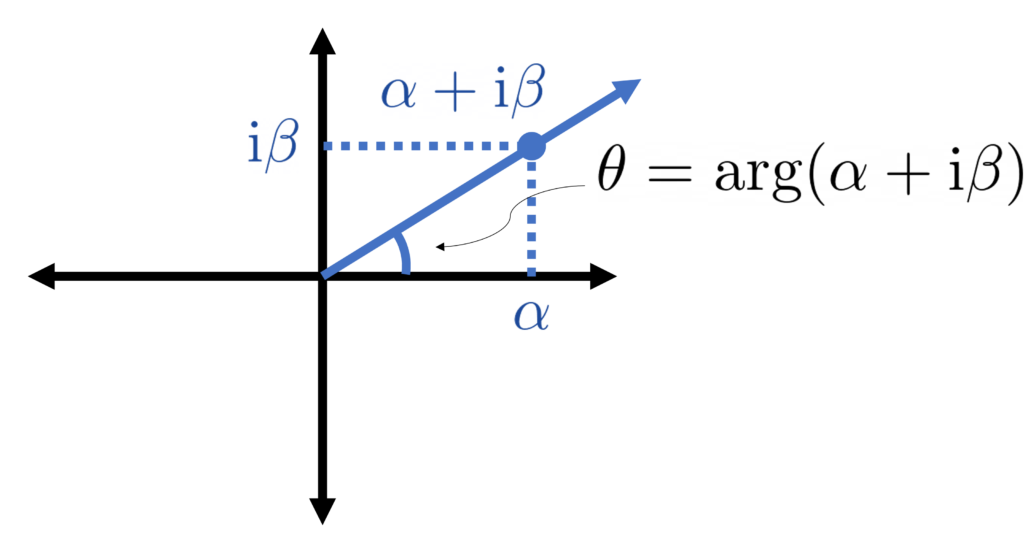


 can be written as the following minimax optimization problem
can be written as the following minimax optimization problem
 of dimension
of dimension  whereas the maximum is taken over all unit vectors
whereas the maximum is taken over all unit vectors 


 .
.![[0,\pi]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-049f2026ce1810bcad11310197f7804b_l3.png "Rendered by QuickLaTeX.com") , this means that the eigenvalues
, this means that the eigenvalues  are now in increasing order, in contrast to our convention from earlier in this section.
are now in increasing order, in contrast to our convention from earlier in this section.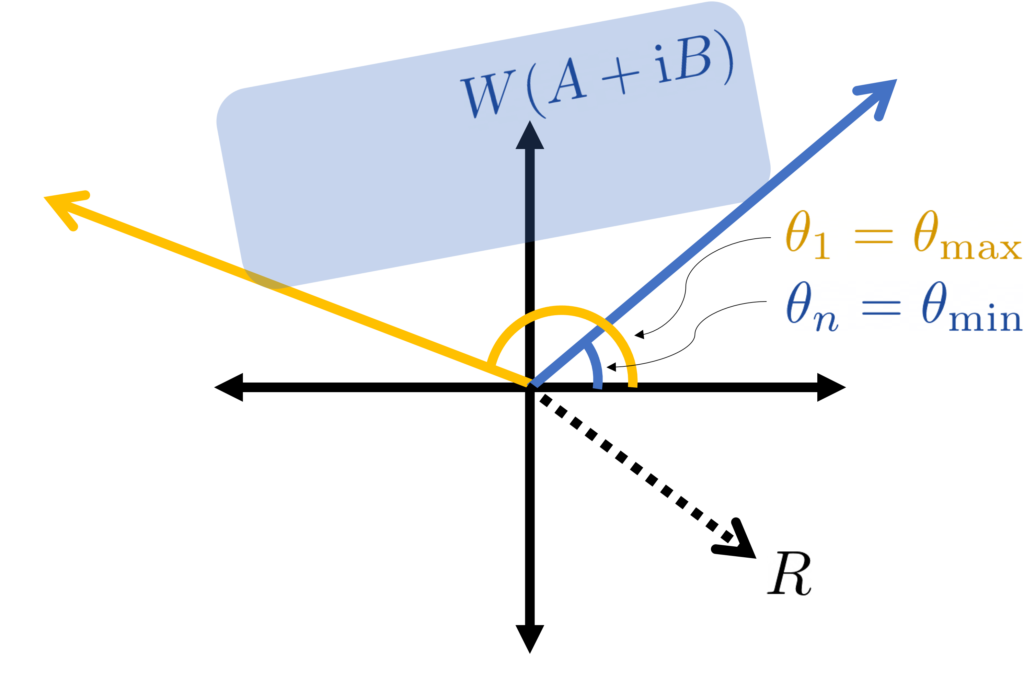
 by the distance between
by the distance between  . But the difference between these two quantities is just
. But the difference between these two quantities is just 
 be? If the vector
be? If the vector  from zero. This quantity has a name:
from zero. This quantity has a name: ) is
) is  .
. .
.
 by more familiar quantities. For instance, once can straightforwardly show the bound
by more familiar quantities. For instance, once can straightforwardly show the bound  , where
, where  is the
is the  denote the eigenangles of the perturbed pair
denote the eigenangles of the perturbed pair  and consider the
and consider the  be the subspace of dimension
be the subspace of dimension 
 . We’re truly toast if the perturbation is large enough to perturb
. We’re truly toast if the perturbation is large enough to perturb  to be equal to zero, so we should assume that
to be equal to zero, so we should assume that  .
.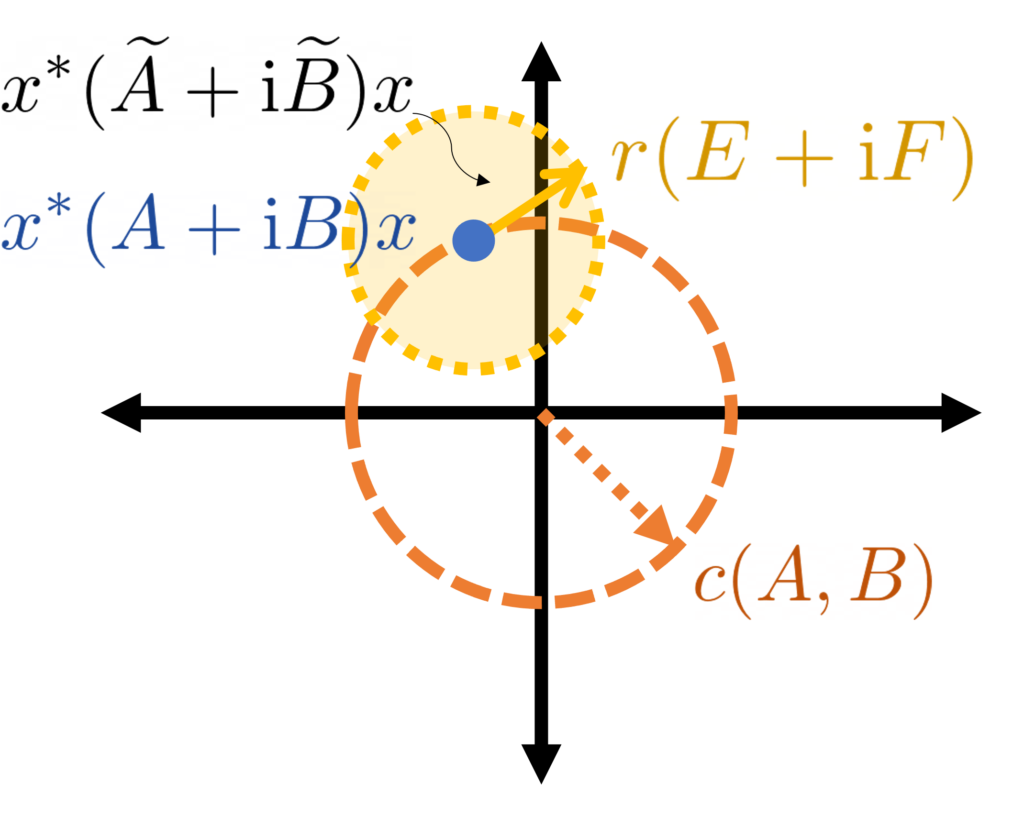
 might lie anywhere in a circle centered at
might lie anywhere in a circle centered at  for every
for every 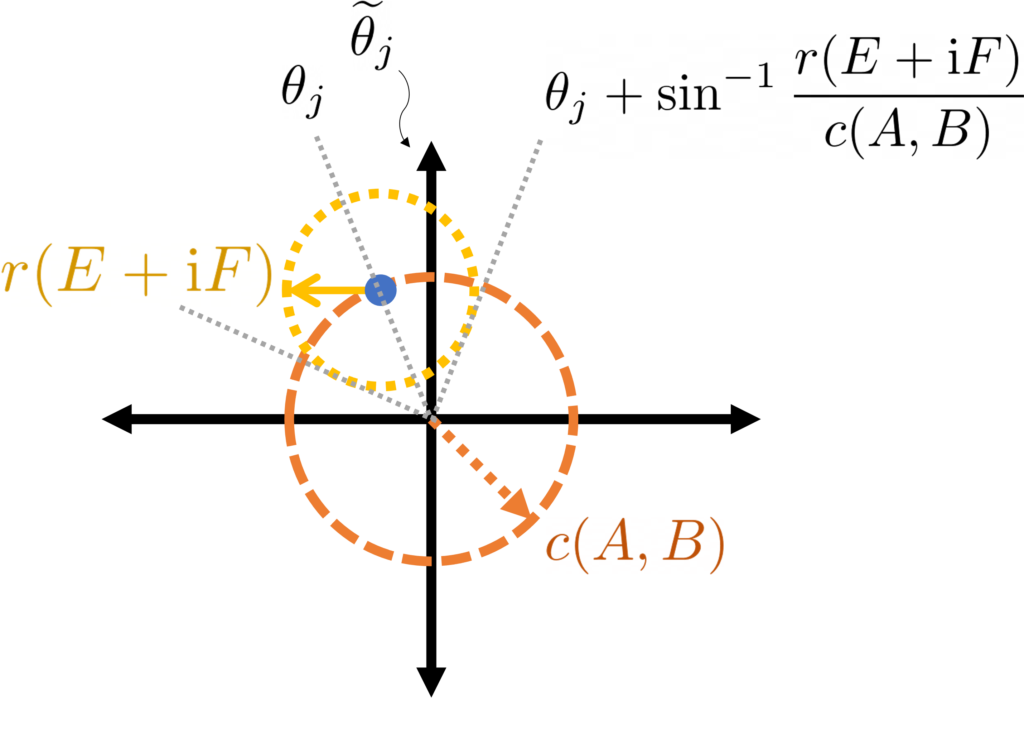
 .
. . An entirely analogous argument using the max-min variational principle (1) proves an identical lower bound, thus showing
. An entirely analogous argument using the max-min variational principle (1) proves an identical lower bound, thus showing

 , is much smaller than the definiteness of the original problem, measured by the Crawford number
, is much smaller than the definiteness of the original problem, measured by the Crawford number  and another hard-to-concisely describe quantity. In many cases, one has nothing better to do than to bound
and another hard-to-concisely describe quantity. In many cases, one has nothing better to do than to bound  , in which case the Crawford number has appeared again.
, in which case the Crawford number has appeared again. . The insight of the Mathias–Li theory is, in some sense, as simple as this: rather than using the fact that
. The insight of the Mathias–Li theory is, in some sense, as simple as this: rather than using the fact that  (as in Stewart’s analysis), use how far
(as in Stewart’s analysis), use how far  —the eigenvectors are thus made “
—the eigenvectors are thus made “ . In this way, just taking the eigenvector
. In this way, just taking the eigenvector  and
and  are diagonal matrices with entries
are diagonal matrices with entries  and
and  respectively. With this in mind, it can make our lives a lot easy to just do a change of variables
respectively. With this in mind, it can make our lives a lot easy to just do a change of variables  and
and  (which in turn sends
(which in turn sends  and
and  ). The change of variables
). The change of variables  . Further, note the following variational characterization of the spectral radius
. Further, note the following variational characterization of the spectral radius  . Plugging these two facts together yields
. Plugging these two facts together yields  .
. , which can be arbitrarily large.
, which can be arbitrarily large. and
and  the
the  and
and  where
where  is the
is the  .
.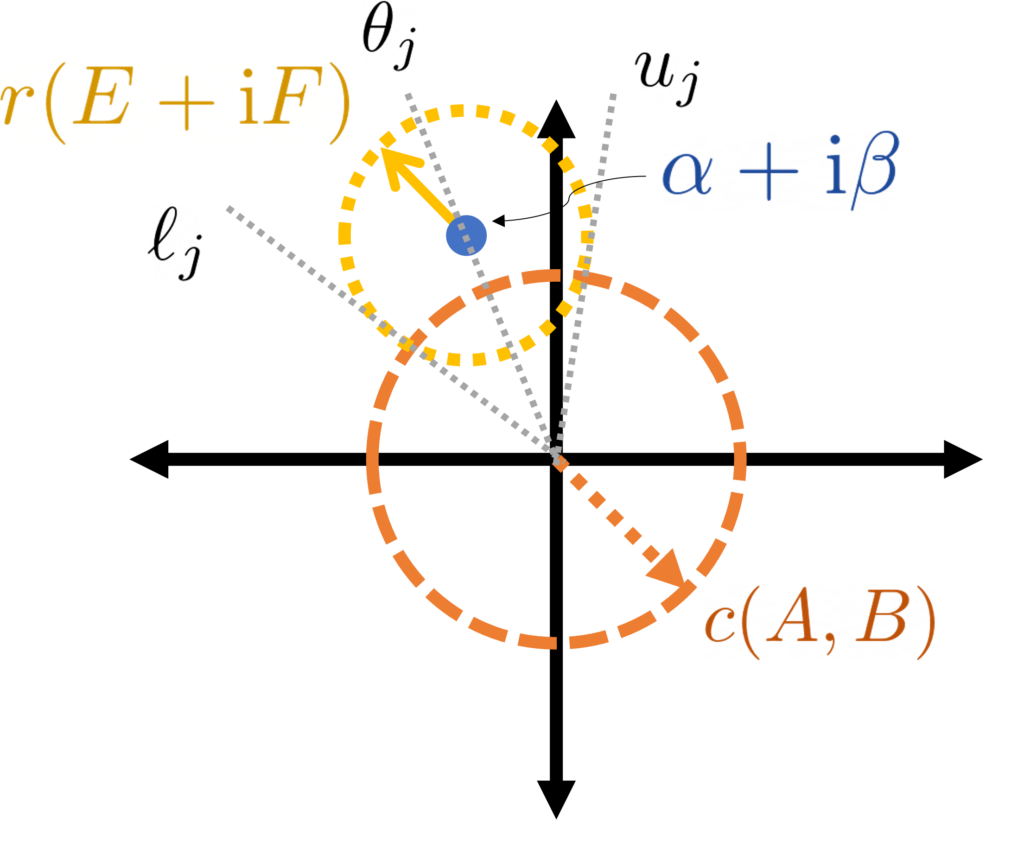
 is the associated eigenangle, then this circle is subtended by arcs with angles
is the associated eigenangle, then this circle is subtended by arcs with angles
 were guaranteed to lie in these arcs (i.e.,
were guaranteed to lie in these arcs (i.e.,  ). Unfortunately this is not necessarily the case. If one
). Unfortunately this is not necessarily the case. If one  . In particular, this means that
. In particular, this means that  in decreasing order
in decreasing order  , we hace
, we hace  . An entirely analogous argument will give an upper bound, yielding
. An entirely analogous argument will give an upper bound, yielding
 . The trick will be to use the max-min characterization (1) with the subspace
. The trick will be to use the max-min characterization (1) with the subspace  . Churning through a couple inequalities in quick fashion,
. Churning through a couple inequalities in quick fashion,
 denotes the
denotes the  , it in particular holds for the set of indices which makes
, it in particular holds for the set of indices which makes  the largest. Thus,
the largest. Thus,  .
. by its minimum over all
by its minimum over all  (and
(and  )
)

 ; as Mathias and Li show
; as Mathias and Li show  .
. and
and  . (The precise instantiation of “sufficiently well-separated” requires some tedious algebra; if you’re interested, see
. (The precise instantiation of “sufficiently well-separated” requires some tedious algebra; if you’re interested, see 
 for a sufficiently small perturbation. In fact, a result of this form is nearly as good as one could hope for.
for a sufficiently small perturbation. In fact, a result of this form is nearly as good as one could hope for. , so we know for sufficiently small perturbations we have
, so we know for sufficiently small perturbations we have  and
and ![[0,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4019f7e1f59924f7600fd76da233c29e_l3.png "Rendered by QuickLaTeX.com") is to pick 100 equispaced points
is to pick 100 equispaced points  on this interval and output the Riemann sum
on this interval and output the Riemann sum  . A randomized algorithm is simply an algorithm which uses, in some form, randomly chosen quantities. For instance, a randomized algorithm
. A randomized algorithm is simply an algorithm which uses, in some form, randomly chosen quantities. For instance, a randomized algorithm uniformly distributed in the interval
uniformly distributed in the interval  . To help to distinguish, an algorithm which does not use randomness is called a deterministic algorithm.
. To help to distinguish, an algorithm which does not use randomness is called a deterministic algorithm. operations
operations  operations
operations integers to sort in increasing order, quicksort works by selecting an element to be the pivot and then divides the elements of the list into groups larger and smaller than the pivot. One then recurses on the two groups, ending up with a sorted list.
integers to sort in increasing order, quicksort works by selecting an element to be the pivot and then divides the elements of the list into groups larger and smaller than the pivot. One then recurses on the two groups, ending up with a sorted list. time, which is asymptotically fast as this problem could possibly be solved since any median-finding algorithm must in general look at the entire list.
time, which is asymptotically fast as this problem could possibly be solved since any median-finding algorithm must in general look at the entire list. expected runtime
expected runtime is the fastest possible runtime for any
is the fastest possible runtime for any  algorithm. If one feeds in a list in a random order, the first-element pivot selection has a
algorithm. If one feeds in a list in a random order, the first-element pivot selection has a  , then we can still come up with an ordering of the input list that makes the algorithm run in time
, then we can still come up with an ordering of the input list that makes the algorithm run in time  .
. over the class of all algorithms
over the class of all algorithms  which solve the problem. (Less formally, we assign each possible algorithm a probability of picking it and pick one subject to these probabilities.) Once your opponent sees your randomized algorithm (equivalently, the distribution
which solve the problem. (Less formally, we assign each possible algorithm a probability of picking it and pick one subject to these probabilities.) Once your opponent sees your randomized algorithm (equivalently, the distribution  and present it to your algorithm.
and present it to your algorithm. with some distribution
with some distribution  to counter their strategy.
to counter their strategy.![\begin{equation*} \mathbb{E}_{I\sim Q} \left[ C(A_{\rm best},I) \right] \le \mathbb{E}_{A\sim P} \left[ \mathbb{E}_{I\sim Q} \left[ C(A, I) \right] \right]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-55ccc5c1f35b1e073bb8ccb0674706e3_l3.png "Rendered by QuickLaTeX.com")
 and
and  denote the
denote the ![\begin{equation*} \mathbb{E}_{A\sim P} \left[ \mathbb{E}_{I\sim Q} \left[ C(A, I) \right] \right] \le \mathbb{E}_{A\sim P} \left[ C(A,I_{\rm worst}) \right]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-08306ab131eefc536da92e60daacd796_l3.png "Rendered by QuickLaTeX.com")
![\begin{equation*} \mathbb{E}_{I\sim Q} \left[ C(A_{\rm best},I) \right] \le \mathbb{E}_{A\sim P} \left[ C(A,I_{\rm worst}) \right]. \end{equation*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-048075ac1ea1ace95f8d86e6c79a981c_l3.png "Rendered by QuickLaTeX.com")
![\max_{\mbox{input distributions } Q} \min_{\mbox{algorithms }A} \mathbb{E}_{I\sim Q} [C(A,I)]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6e5056cc57c21521a44f1bae4fa64f69_l3.png "Rendered by QuickLaTeX.com")
![=\min_{\mbox{randomized algorithms }P} \max_{\mbox{inputs }I}\mathbb{E}_{A\sim P} \left[ C(A,I) \right].](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9651554bb21521de67225ea9a22d15e6_l3.png "Rendered by QuickLaTeX.com") This nontrivial fact is a consequence of the full power of the
This nontrivial fact is a consequence of the full power of the  . If one in addition allows randomness, we call this
. If one in addition allows randomness, we call this  .
. .
. : all problems that can be tractably solved with randomness can tractably be solved without. There is
: all problems that can be tractably solved with randomness can tractably be solved without. There is 
 .
. , where
, where  and
and  th entry
th entry  .
. being replaced by
being replaced by  . The columns of the matrices
. The columns of the matrices  and
and  .
. and
and  . This follows from the calculations
. This follows from the calculations  and
and  . In other words, the nonzero singular values of
. In other words, the nonzero singular values of  or
or  . (These formulas are valid for invertible square matrices
. (These formulas are valid for invertible square matrices  of
of  and
and  may not be orthogonal matrices with, for example,
may not be orthogonal matrices with, for example,  not even close to the identity matrix. One can, of course, orthogonalize the computed
not even close to the identity matrix. One can, of course, orthogonalize the computed 
 are precisely plus-or-minus the singular values of
are precisely plus-or-minus the singular values of 
 and thus
and thus  for
for  account for all the nonzero eigenvalues of
account for all the nonzero eigenvalues of  is linear.
is linear. , an
, an  (respectively,
(respectively,  ), then
), then