I am excited to share that my paper Does block size matter in randomized block Krylov low-rank approximation? has recently been released on arXiv. In that paper, we study the randomized block Krylov iteration (RBKI) algorithm for low-rank approximation. Existing results show that RBKI is efficient at producing rank- approximations with a large block size of or a small block size of
approximations with a large block size of or a small block size of  , but these results give poor results for intermediate block sizes
, but these results give poor results for intermediate block sizes  . But often these intermediate block sizes are the most efficient in practice. In our paper, we close this theoretical gap, showing RBKI is efficient for any block size
. But often these intermediate block sizes are the most efficient in practice. In our paper, we close this theoretical gap, showing RBKI is efficient for any block size  . Check out the paper for details!
. Check out the paper for details!
In our paper, the core technical challenge is understanding the condition number of a random block Krylov matrix of the form
![\[K = \begin{bmatrix} G & AG & \cdots & A^{t-1}G \end{bmatrix},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a6ec37fe54d09f2ee9f6f2c648dafd1_l3.png "Rendered by QuickLaTeX.com")
 is an
is an  real symmetric positive semidefinite matrix and
real symmetric positive semidefinite matrix and  is an
is an  random Gaussian matrix. Proving this result was not easy, and our proof required several ingredients. In this post, I want to talk about just one of them: Gautschi’s bound on the conditioning of Vandermonde matrices. (Check out Gautschi’s original paper here.)
random Gaussian matrix. Proving this result was not easy, and our proof required several ingredients. In this post, I want to talk about just one of them: Gautschi’s bound on the conditioning of Vandermonde matrices. (Check out Gautschi’s original paper here.)
Evaluating a Polynomial and Vandermonde Matrices
Let us begin with one the humblest but most important characters in mathematics, the univariate polynomial
![\[p_a(u) = a_1 + a_2 u+ \cdots + a_t u^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ba898ff67858e9c5a76463593337b542_l3.png "Rendered by QuickLaTeX.com")
 and have written the polynomial
and have written the polynomial  parametrically in terms of its vector of coefficients
parametrically in terms of its vector of coefficients  . The polynomials of degree at most form a linear space of dimension
. The polynomials of degree at most form a linear space of dimension  , and the monomials
, and the monomials  provide a basis for this space. In this post, we will permit the coefficients
provide a basis for this space. In this post, we will permit the coefficients  to be complex numbers.
to be complex numbers.
Given a polynomial, we often wish to evaluate it at a set of inputs. Specifically, let  be
be  (distinct) input locations. If we evaluate
(distinct) input locations. If we evaluate  at each number, we obtain a list of (output) values, which we denote by
at each number, we obtain a list of (output) values, which we denote by  of (distinct) values, each of which given by the formula
of (distinct) values, each of which given by the formula
![\[p_a(\lambda_i) = \sum_{j=1}^t \lambda_i^{j-1} a_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eaaf53acb1f30e1ced80af4704800e02_l3.png "Rendered by QuickLaTeX.com")
 are a nonlinear function of the input values
are a nonlinear function of the input values  but a linear function of the coefficients
but a linear function of the coefficients  . We may call the mapping
. We may call the mapping  the coefficients-to-values map.
the coefficients-to-values map.
Every linear transformation between vectors can be realized as a matrix–vector product, and the matrix for the coefficients-to-values map is called a Vandermonde matrix  . It is given by the formula
. It is given by the formula
![\[V= \begin{bmatrix} 1 & \lambda_1 & \lambda_1^2 & \cdots & \lambda_1^{t-1} \\ 1 & \lambda_2 & \lambda_2^2 & \cdots & \lambda_2^{t-1} \\ 1 & \lambda_3 & \lambda_3^2 & \cdots & \lambda_3^{t-1} \\ \vdots & \vdots & \vdots & \ddots & \vdots \\ 1 & \lambda_s & \lambda_s^2 & \cdots & \lambda_s^{t-1} \end{bmatrix} \in \complex^{s\times t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d100f803f43a4910ba6c03d2edde384_l3.png "Rendered by QuickLaTeX.com")
 .
.
Interpolating by a Polynomial and Inverting a Vandermonde Matrix
Going forward, let us set  so the number of locations
so the number of locations  equals the number of coefficients . The Vandermonde matrix maps the vector of coefficients to the vector of values
equals the number of coefficients . The Vandermonde matrix maps the vector of coefficients to the vector of values  . Its inverse
. Its inverse  maps a set of values to a set of coefficients defining a polynomial which interpolates the values :
maps a set of values to a set of coefficients defining a polynomial which interpolates the values :
![\[p_a(\lambda_i) = f_i \quad \text{for } i =1,\ldots,t .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0cab3d96a2006c60b95e665b3c9afc97_l3.png "Rendered by QuickLaTeX.com")
To solve the polynomial interpolation problem with Vandermonde matrices, we can do the following. Given values , we first solve the linear system of equations  , obtaining a vector of coefficients
, obtaining a vector of coefficients  . Then, define the interpolating polynomial
. Then, define the interpolating polynomial
(1) ![\[q(u) = a_1 + a_2 u + \cdots + a_t u^{t-1} \quad \text{with } a = V^{-1} f. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b44af0dbb2f4e6b3c74a40b23d074f68_l3.png "Rendered by QuickLaTeX.com")
 interpolates the values at the locations
interpolates the values at the locations  ,
,  .
.
Lagrange Interpolation
Inverting a the Vandermonde matrix is one way to solve the polynomial interpolation problem, but the polynomial interpolation can also be solved directly. To do so, first notice that we can construct a special polynomial  that is zero at the locations
that is zero at the locations  but nonzero at the first location
but nonzero at the first location  . (Remember that we have assumed that
. (Remember that we have assumed that  are distinct.) Further, by rescaling this polynomial to
are distinct.) Further, by rescaling this polynomial to
![\[\ell_1(u) = \frac{(u - \lambda_2)(u - \lambda_3)\cdots(u-\lambda_t)}{(\lambda_1 - \lambda_2)(\lambda_1 - \lambda_3)\cdots(\lambda_1-\lambda_t)},\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-174996c439ec13555388925748a3269e_l3.png "Rendered by QuickLaTeX.com")
can be set to . Likewise, for each  , we can define a similar polynomial
, we can define a similar polynomial (2) ![\[\ell_i(u) = \frac{(u - \lambda_1)\cdots(u-\lambda_{i-1}) (u-\lambda_{i+1})\cdots(u-\lambda_t)}{(\lambda_i - \lambda_1)\cdots(\lambda_i-\lambda_{i-1}) (\lambda_i-\lambda_{i+1})\cdots(\lambda_i-\lambda_t)}, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5453dea91bda2e744ca98b5dc0ae9790_l3.png "Rendered by QuickLaTeX.com")
at and zero at  for
for  . Using the Dirac delta symbol, we may write
. Using the Dirac delta symbol, we may write ![\[\ell_i(\lambda_j) = \delta_{ij} = \begin{cases} 1, & i = j, \\ 0, & i\ne j. \end{cases}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-64c4effeb0f12225a76059d89ccce692_l3.png "Rendered by QuickLaTeX.com")
 are called the Lagrange polynomials of the locations
are called the Lagrange polynomials of the locations  . Below is an interactive illustration of the second Lagrange polynomial
. Below is an interactive illustration of the second Lagrange polynomial  associated with the points
associated with the points  (with
(with  ).
).
With the Lagrange polynomials in hand, the polynomial interpolation problem is easy. To obtain a polynomial whose values are , simply multiply each Lagrange polynomial by the value  and sum up, obtaining
and sum up, obtaining
![\[q(u) = \sum_{i=1}^t f_i \ell_i(u).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c1c5043718b5df795a883a6a60d26aaf_l3.png "Rendered by QuickLaTeX.com")
interpolates the values . Indeed, (3) ![\[q(\lambda_j) = \sum_{i=1}^t f_i \ell_i(\lambda_j) = \sum_{i=1}^t f_i \delta_{ij} = f_j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-90032b377ebe250ef36dccba2186b6ce_l3.png "Rendered by QuickLaTeX.com")
From Lagrange to Vandermonde via Elementary Symmetric Polynomials
We now have two ways of solving the polynomial interpolation problem, the Vandermonde way (1) and the Lagrange way (3). Ultimately, the difference between these formulas is one of basis: The Vandermonde formula (1) expresses the interpolating polynomial as a linear combination of monomials and the Lagrange formula (3) expresses as a linear combination of the Lagrange polynomials  . To convert between these formulas, we just need to express the Lagrange polynomial basis in the monomial basis.
. To convert between these formulas, we just need to express the Lagrange polynomial basis in the monomial basis.
To do so, let us examine the Lagrange polynomials more closely. Consider first the case  , and consider the fourth unnormalized Lagrange polynomial
, and consider the fourth unnormalized Lagrange polynomial
![\[(u - \lambda_1) (u - \lambda_2) (u - \lambda_3).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d3fe2471c7c0557476992f6f7a1361cf_l3.png "Rendered by QuickLaTeX.com")
 , we obtain the expression
, we obtain the expression ![\[(u - \lambda_1) (u - \lambda_2) (u - \lambda_3) = u^3 - (\lambda_1 + \lambda_2 + \lambda_3) u^2 + (\lambda_1\lambda_2 + \lambda_1\lambda_3 + \lambda_2\lambda_3)u - \lambda_1\lambda_2\lambda_3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ad4d01202c2b9a69e395412829cc7fed_l3.png "Rendered by QuickLaTeX.com")
 , we recognize some pretty distinctive expressions involving the ‘s:
, we recognize some pretty distinctive expressions involving the ‘s:

Indeed, these expressions are special. Up to a plus-or-minus sign, they are called the elementary symmetric polynomials of the locations . Specifically, given numbers  , the th elementary symmetric polynomial
, the th elementary symmetric polynomial  is defined as the sum of all products
is defined as the sum of all products  of values, i.e.,
of values, i.e.,
![\[e_k(\mu_1,\ldots,\mu_{t-1}) = \sum_{i_1 < i_2 < \cdots < i_k} \mu_{i_1}\mu_{i_2}\cdots \mu_{i_k}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-49b00bf8f1a0f6fc399e029516a4682e_l3.png "Rendered by QuickLaTeX.com")
by convention.
The elementary symmetric polynomials appear all the time in mathematics. In particular, they are the coefficients of the characteristic polynomial of a matrix and feature heavily in the theory of determinantal point processes. For our purposes, the key observation will be that the elementary symmetric polynomials appear whenever one expands out an expression like  :
:
Lemma 1 (Expanding a product of linear functions). It holds that
![\[(u + \mu_1) (u + \mu_2) \cdots (u+\mu_k) = \sum_{j=0}^k e_{k-j}(\mu_1,\ldots,\mu_k) u^j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fe26dd7ba2e215fba4fea5cd28147cb2_l3.png "Rendered by QuickLaTeX.com")
Using this fact, we obtain an expression for the Lagrange polynomials in the monomial basis. Let  denote the list of locations without . Then the th Lagrange polynomial is given by
denote the list of locations without . Then the th Lagrange polynomial is given by
![\[\ell_i(u) = \frac{\sum_{j=1}^t e_{t-j}(-\lambda_{-i})u^{j-1}}{\prod_{k\ne i} (\lambda_i - \lambda_k)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-15d6cb602dcf916e14363b25a013870d_l3.png "Rendered by QuickLaTeX.com")
Indeed, we can write the interpolating polynomial as
![\[q(u) = \sum_{i=1}^t f_i\ell_i(u) = \sum_{i=1}^t \sum_{j=1}^t f_i \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)} u^{j-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-17012971c8949a309b08ae230bf7fa94_l3.png "Rendered by QuickLaTeX.com")
![\[q(u) = \sum_{j=1}^t \sum_{i=1}^t f_i \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)} u^{j-1} = \sum_{j=1}^t \left(\sum_{i=1}^t \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}\cdot f_i \right) u^{j-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-10960bce48c6a124c253db4c1272f2e2_l3.png "Rendered by QuickLaTeX.com")
![\[a_j = \sum_{i=1}^t \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}\cdot f_i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2bb3d1280c99ca124084f031ed343679_l3.png "Rendered by QuickLaTeX.com")
. Therefore, we conclude that the entries of the inverse-Vandermonde matrix are (4) ![\[(V^{-1})_{ji} = \frac{e_{t-j}(-\lambda_{-i})}{\prod_{k\ne i} (\lambda_i - \lambda_k)}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c3d618b9ee7195d040d1875f76fb1d67_l3.png "Rendered by QuickLaTeX.com")
Vandermonde Matrices are Merely Exponentially Ill-Conditioned
Vandermonde matrices are notoriously ill-conditioned, meaning that small changes to the values can cause large changes to the coefficients . On its face, this might seem like the problem of polynomial interpolation itself is ill-conditioned, but this is too hasty a conclusion. After all, it is only the mapping from values to coefficients in the monomial basis that is ill-conditioned. Fortunately, there are much better, more numerically stable bases for representing a polynomial like the Chebyshev polynomials.
But these more stable methods of polynomial interpolation and approximation are not the subject of this post: Here, our task is to will be to characterize just how ill-conditioned the computation of is. To characterize this ill-conditioning, we will utilize the condition number of the matrix . Given a norm  , the condition number of is defined to be
, the condition number of is defined to be
![\[\kappa_{\uinorm{\cdot}}(V) = \uinorm{V} \uinorm{\smash{V^{-1}}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cf4da36f5f2ce121d2d2512423310221_l3.png "Rendered by QuickLaTeX.com")
 is chosen to be the (operator) –norm, defined as
is chosen to be the (operator) –norm, defined as ![\[\norm{A}_1= \max_{x \ne 0} \frac{\norm{Ax}_1}{\norm{x}_1} \quad \text{where } \norm{x}_1 = \sum_i |x_i|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-beb68f2e11bc5c6e30b06fec3b886ff3_l3.png "Rendered by QuickLaTeX.com")
-norm has a simple characterization: It is the maximum sum of the absolute values of the entries in any column (5) ![\[\norm{A}_\infty = \max_j \sum_i |A_{ij}|. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-936a7309e99d6c03a128b3a7771c88df_l3.png "Rendered by QuickLaTeX.com")
-norm condition number  .
.
Bounding  is straightforward. Indeed, setting
is straightforward. Indeed, setting  and using (5), we compute
and using (5), we compute
![\[\norm{V}_1= \max_{1\le j \le t} \sum_{i=1}^t |\lambda_i|^{j-1} \le \max_{1\le j \le t} tM^{j-1} = t\max\{1,M^{t-1}\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ec5f488cb8233df69dd22a00e9bae784_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{V}_1\le t(1+M)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8cecac332b3fcbe899304e963ec1fe47_l3.png "Rendered by QuickLaTeX.com")
The harder task is bounding  . Fortunately, we have already done most of the hard work needed to bound this quantity. Using our expression (4) for the entries of and using the formula (5) for the -norm, we have
. Fortunately, we have already done most of the hard work needed to bound this quantity. Using our expression (4) for the entries of and using the formula (5) for the -norm, we have
![\[\norm{\smash{V^{-1}}}_1= \max_{1\le j \le t} \sum_{i=1}^t |(V^{-1})_{ij}| = \max_{1\le j \le t}\frac{ \sum_{i=1}^t |e_{t-i}(-\lambda_{-j})|}{\prod_{k\ne j} |\lambda_j- \lambda_k|}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b431915bf4dd9e0a3087cd91f5e3533a_l3.png "Rendered by QuickLaTeX.com")
![\[|e_k(\mu_1,\ldots,\mu_s)| \le e_k(|\mu_1|,\ldots,|\mu_s|).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-73a7d10b6aadf0271cb538d648049f13_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain ![\[\norm{\smash{V^{-1}}}_1\le \max_{1\le j \le t}\frac{ \sum_{i=1}^t e_{t-i}(|\lambda_{-j}|)}{\prod_{k\ne j} |\lambda_j- \lambda_k|}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-34c4b502a43c91513781e51c25190ad1_l3.png "Rendered by QuickLaTeX.com")
 to obtain the expression
to obtain the expression (6) ![\[\norm{\smash{V^{-1}}}_1\le \max_{1\le j \le t}\frac{ \prod_{k\ne j} (1 + |\lambda_k|)}{\prod_{k\ne j} |\lambda_j- \lambda_k|}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9fee19675094e57026eaf8571e060317_l3.png "Rendered by QuickLaTeX.com")
Often, it is helpful to simply Gautschi’s bound a bit. Setting as above, the numerator is bounded as  . To bound the denominator, let
. To bound the denominator, let  be the smallest distance between two locations. Using
be the smallest distance between two locations. Using  and
and  , we can weaken the bound (6) to obtain
, we can weaken the bound (6) to obtain
![\[\norm{\smash{V^{-1}}}_1\le \left(\frac{1+M}{\mathrm{gap}}\right)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4df225e8dea0e69df68907fc81ab32e6_l3.png "Rendered by QuickLaTeX.com")
 from above, we obtain a bound on the condition number
from above, we obtain a bound on the condition number ![\[\kappa_1(V) \le t \left( \frac{(1+M)^2}{\mathrm{gap}} \right)^{t-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a88bac288484194059179a7e854bc368_l3.png "Rendered by QuickLaTeX.com")
Theorem 2 (Gautschi’s bound, simplified). Introduce
. Then
and
![\[\norm{\smash{V^{-1}}}_1\le \left(\frac{1+M}{\mathrm{gap}}\right)^{t-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-933010efafd6e0336cdf5b4071c2c90b_l3.png "Rendered by QuickLaTeX.com")
Gautschi’s bound suggests that Vandermonde matrices can be very ill-conditioned, which is disappointing. But Gautschi’s bound also shows that Vandermonde matrices are merely exponentially ill-conditioned—that is, they are not worse than exponentially conditioned. The fact that Vandermonde matrices are only exponentially ill-conditioned plays a crucial role in our analysis of randomized block Krylov iteration.
Gautschi’s Bound as a Robust Version of the Fundamental Theorem of Algebra
The fundamental theorem of algebra states that a degree- polynomial has precisely roots. Consequently, at locations, it must be nonzero at least one. But how nonzero must the polynomial be at that one location? How large must it be? On this subject, the fundamental theorem of algebra is moot. However, Gautschi’s bound provides an answer.
polynomial has precisely roots. Consequently, at locations, it must be nonzero at least one. But how nonzero must the polynomial be at that one location? How large must it be? On this subject, the fundamental theorem of algebra is moot. However, Gautschi’s bound provides an answer.
To answer this question, we ask: What is the minimum possible size  of the values
of the values  ? Well, if we set all the coefficients
? Well, if we set all the coefficients  to zero, then
to zero, then  as well. So to avoid this trivial case, we should enforce a normalization condition on the coefficient vector , say
as well. So to avoid this trivial case, we should enforce a normalization condition on the coefficient vector , say  . With this setting, we are ready to compute. Begin by observing that
. With this setting, we are ready to compute. Begin by observing that
![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \min_{a\ne 0} \frac{\norm{Va}_1}{\norm{a}_1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3a21773d318eeb447cc3deec7d2ec9e1_l3.png "Rendered by QuickLaTeX.com")
 , obtaining
, obtaining ![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \min_{a\ne 0} \frac{\norm{Va}_1}{\norm{a}_1} = \min_{f\ne 0} \frac{\norm{f}_1}{\norm{\smash{V^{-1}f}}_1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e223271ff5f99accd84933f4936864bd_l3.png "Rendered by QuickLaTeX.com")
![\[\left(\min_{\norm{a}_1 = 1} \norm{Va}_1\right)^{-1} = \max_{f\ne 0} \frac{\norm{\smash{V^{-1}f}}_1}{\norm{f}_1} = \norm{\smash{V^{-1}}}_1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-745fdb308eb43cd71eb1fda9a7b16762_l3.png "Rendered by QuickLaTeX.com")
![\[\min_{\norm{a}_1 = 1} \norm{Va}_1 = \norm{\smash{V^{-1}}}_1^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-145e10d104c261b8eba6886d9e5a88d0_l3.png "Rendered by QuickLaTeX.com")
Proposition 3 (Minimum stretch). For vector norm
![\[\min_{\uinorm{v} = 1} \uinorm{Av} = \min_{v\ne 0} \frac{\uinorm{Av}}{\uinorm{v}} = \uinorm{\smash{A^{-1}}}^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c07d1dd4195814db2ab892e8587fb9b4_l3.png "Rendered by QuickLaTeX.com")
Using this result, we obtain the lower bound  on the values of a polynomial with coefficients . Combining with Gautschi’s bound gives the following robust version of the fundamental theorem of algebra:
on the values of a polynomial with coefficients . Combining with Gautschi’s bound gives the following robust version of the fundamental theorem of algebra:
Theorem 4 (Robust fundamental theorem of algebra). Fix a polynomial
and locations
![\[|p(\lambda_1)| + \cdots + |p(\lambda_t)| \ge \left(\frac{\mathrm{gap}}{1+M}\right)^{t-1} (|a_1| + \cdots + |a_t|).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f011798a0af29d677a21bf1d0fb00152_l3.png "Rendered by QuickLaTeX.com")
Thus, at locations, a degree- polynomial must be nonzero at least one point. In fact, the sum of the values at these locations must be no worse than exponentially small in .
 , whose dimensions will be
, whose dimensions will be  throughout this post. Beginning from a trivial initial solution
throughout this post. Beginning from a trivial initial solution  , the method works by repeating the following two steps for
, the method works by repeating the following two steps for  :
: of
of ![\[\prob\{ i_t = j\} = \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-81c1ff56386b62fc105c714e21f13f5c_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  th row of
th row of  onto the solution space of the equation
onto the solution space of the equation  , obtaining
, obtaining  .
. satisfying
satisfying  ), RK is
), RK is ![\[\expect\left[ \norm{x_t - x_\star}^2 \right] \le (1 - \kappa_{\rm dem}^{-2})^t \norm{x_\star}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-02d23e54a2a02cf261b7ab8afc0f4b90_l3.png "Rendered by QuickLaTeX.com")
![\[\kappa_{\rm dem} = \frac{\norm{A}_{\rm F}}{\sigma_{\rm min}(A)} = \sqrt{\sum_i \left(\frac{\sigma_i(A)}{\sigma_{\rm min}(A)}\right)^2} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a01bce740f28deced2d1a8b4e6e1002_l3.png "Rendered by QuickLaTeX.com")
 are the
are the  , where
, where  is the
is the  is the
is the  , so it takes roughly
, so it takes roughly  row accesses to reduce the error by a constant factor. Compare this to gradient descent, which requires
row accesses to reduce the error by a constant factor. Compare this to gradient descent, which requires  row accesses.
row accesses. , which can be expressed using the
, which can be expressed using the  .
.![\[x_\star = \operatorname{argmin}_{x \in \real^d} \norm{b - Ax}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9052e54b28c6061d7a56c05ca254f154_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\mathbb{E}[x_t] - x_\star}^2 \le (1 - \kappa_{\rm dem}^{-2})^{2t} \norm{x_\star}^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a54409c570b03a8e2e3607c316004f4d_l3.png "Rendered by QuickLaTeX.com")
 .
. . However, we can improve the quality of the approximate solution
. However, we can improve the quality of the approximate solution  which could then be averaged together. This approach is inefficient as each solution
which could then be averaged together. This approach is inefficient as each solution  is computed separately.
is computed separately. , chosen so that the bias
, chosen so that the bias ![\norm{\expect[x_{t_{\rm b}}] - x_\star}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-03b1f60b09d6e66904287aa5e6cd6009_l3.png "Rendered by QuickLaTeX.com") is small. For each
is small. For each  ,
,  is a nearly unbiased approximation to the least-squares solution
is a nearly unbiased approximation to the least-squares solution ![\[\overline{x}_t = \frac{x_{t_{\rm b} +1} + x_{t_{\rm b}+2} + \cdots + x_t}{t-t_{\rm b}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-53375d695acfc74d6ea111d3a1b32d29_l3.png "Rendered by QuickLaTeX.com")
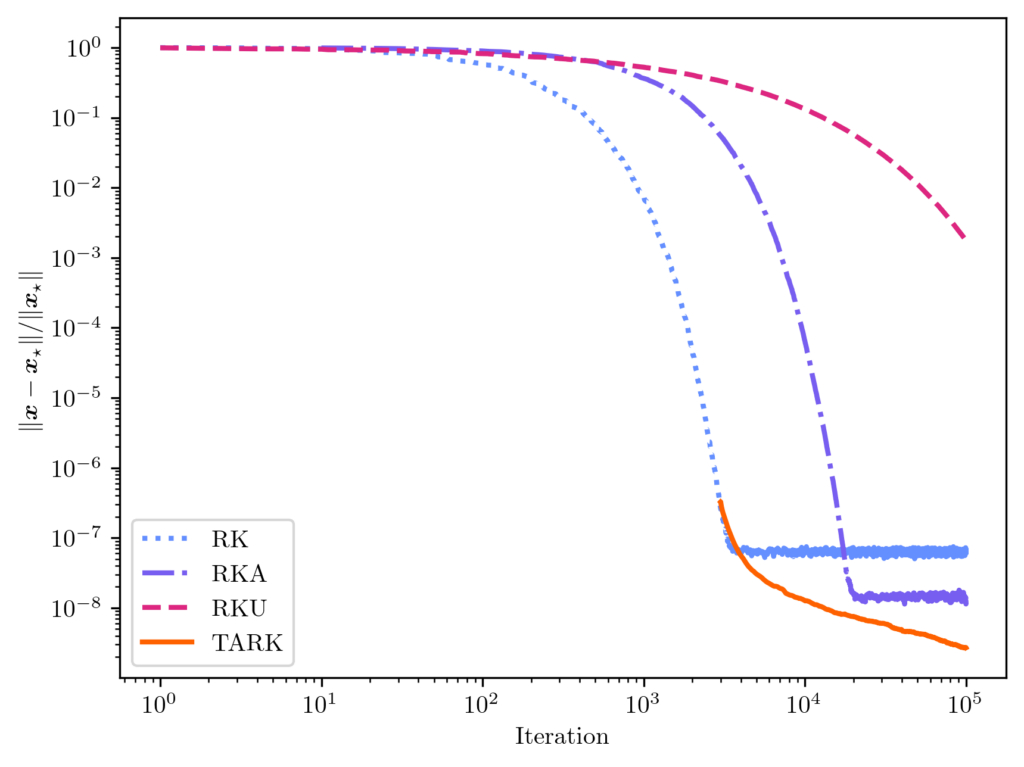
 is the tail-averaged randomized Kaczmarz (TARK) estimator. By Theorem 1, we know the TARK estimator has an exponentially small bias:
is the tail-averaged randomized Kaczmarz (TARK) estimator. By Theorem 1, we know the TARK estimator has an exponentially small bias: ![\[\norm{\expect[\overline{x}_t] - x_\star} \le (1 - \kappa_{\rm dem}^{-2})^{2(t_{\rm b}+1)} \norm{x_\star}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-89806db87c210bfdd9cbd0e800d65e85_l3.png "Rendered by QuickLaTeX.com")
![\[\expect [\norm{\overline{x}_t - x_\star}^2] \le (1-\kappa_{\rm dem}^{-2})^{t_{\rm b}+1} \norm{x_\star}^2 + \frac{2\kappa_{\rm dem}^4}{t-t_{\rm b}} \frac{\norm{b-Ax_\star}^2}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d30cb224e4284f0840a46a9757196abf_l3.png "Rendered by QuickLaTeX.com")
 ;
;  . We found this underrelaxation parameter to lead to a smaller error than the other popular underrelaxation parameter schedule
. We found this underrelaxation parameter to lead to a smaller error than the other popular underrelaxation parameter schedule  .
.
![\[\prob \{ i_t = j \} = \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2} \eqqcolon p^{\rm RK}_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-072dd81a7e854128bf0258a1836003de_l3.png "Rendered by QuickLaTeX.com")
 .
.
 .
. :
: ![\[\prob \{i_t = j\} = p_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-95b2f6411ef8d9b8c9b5b61acf6bc928_l3.png "Rendered by QuickLaTeX.com")
![\[D \coloneqq \diag\left( \sqrt{\frac{p_j}{p_j^{\rm RK}}} : j =1,\ldots,n\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bc930770f7b4e20afffe903a4ab372ba_l3.png "Rendered by QuickLaTeX.com")
 is equivalent to the standard RK algorithm run on the diagonally reweighted least-squares problem
is equivalent to the standard RK algorithm run on the diagonally reweighted least-squares problem ![\[x_{\rm weighted} = \argmin_{x\in\real^d} \norm{Db-(DA)x}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3e1818420a485f35a47361344560d2b1_l3.png "Rendered by QuickLaTeX.com")
 rather than the original least-squares solution
rather than the original least-squares solution  [mfn}Note that, for this experiment we represent the polynomial
[mfn}Note that, for this experiment we represent the polynomial  using its monomial coefficients
using its monomial coefficients  , which has
, which has  equispaced points. We compare the unweighted least-squares solution (orange solid curve) to the weighted least-squares solution using uniform RK weights
equispaced points. We compare the unweighted least-squares solution (orange solid curve) to the weighted least-squares solution using uniform RK weights  (blue dash-dotted curve). These two curves differ meaningfully, with the weighted least-squares solution having higher error at the ends of the interval but more accuracy in the middle. These differences can be explained looking at the weights (diagonal entries of
(blue dash-dotted curve). These two curves differ meaningfully, with the weighted least-squares solution having higher error at the ends of the interval but more accuracy in the middle. These differences can be explained looking at the weights (diagonal entries of  , grey dotted curve), which are lower at the ends of the interval than in the center.
, grey dotted curve), which are lower at the ends of the interval than in the center.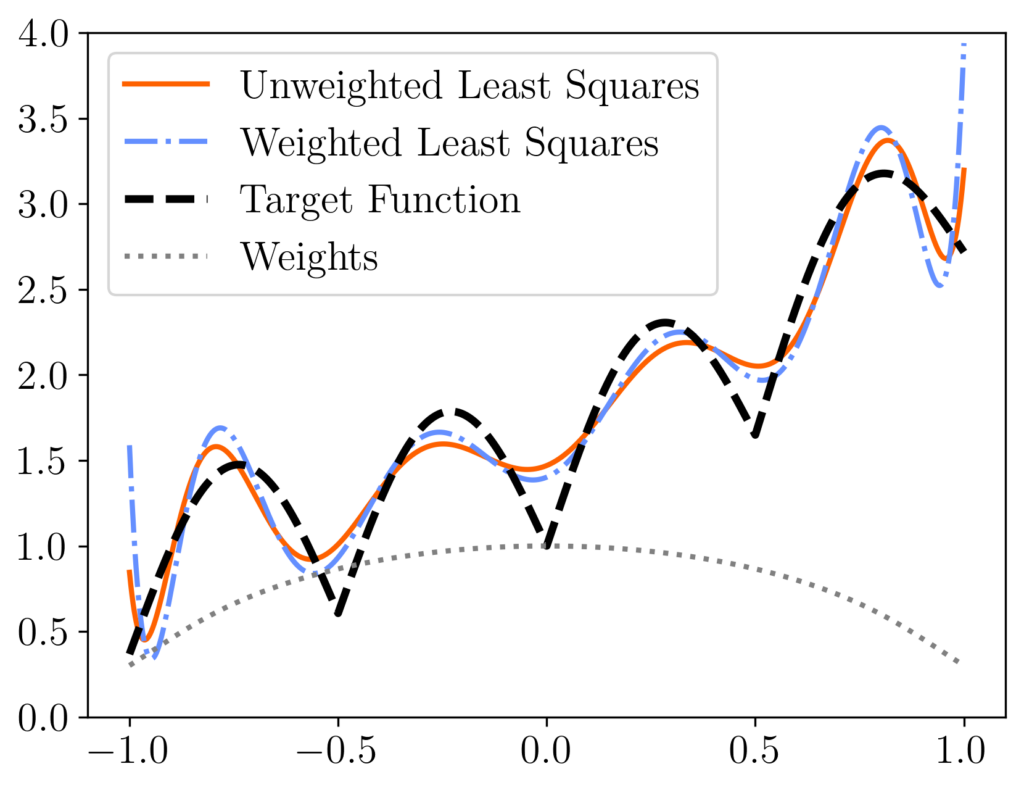
![\[x_{t+1} = \left( I - \frac{a_{i_t}^{\vphantom{\top}}a_{i_t}^\top}{\norm{a_{i_t}}^2} \right)x_t + \frac{b_{i_t}a_{i_t}}{\norm{a_{i_t}}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cccfb54762bb990499c96331e7019309_l3.png "Rendered by QuickLaTeX.com")
 denote the
denote the ![\begin{align*}\expect_{i_t}[x_{t+1}] &= \sum_{j=1}^n \left[\left( I - \frac{a_j^{\vphantom{\top}} a_j^\top}{\norm{a_j}^2} \right)x_t + \frac{b_ja_j}{\norm{a_j}^2}\right] \prob\{i_t=j\}\\ &=\sum_{j=1}^n \left[\left( \frac{\norm{a_j}^2}{\norm{A}_{\rm F}^2}I - \frac{a_j^{\vphantom{\top}} a_j^\top}{\norm{A}_{\rm F}^2} \right)x_t + \frac{b_ja_j}{\norm{A}_{\rm F}^2}\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0a7f7b3bfad51b7094348db94d2d4254_l3.png "Rendered by QuickLaTeX.com")
 and
and  directly. Therefore, we obtain
directly. Therefore, we obtain ![\[\expect_{i_t}[x_{t+1}] = \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right) x_t + \frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-de4ded17daf001dde92c26025e9c14f3_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[x_{t+1}] = \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right) \expect[x_t] + \frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-24c5bfe6c1ea70293055deb68a3df117_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain ![\[\expect[x_t] = \left[\sum_{i=0}^{t-1} \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i \right]\cdot\frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-eb47c96091b1187b53c318589b696693_l3.png "Rendered by QuickLaTeX.com")
![\expect[x_t]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fa98c6708608ae4bf5513cf53e74597c_l3.png "Rendered by QuickLaTeX.com") using a matrix
using a matrix  satisfies the formula
satisfies the formula ![\[\sum_{i=0}^\infty y^i = (1-y)^{-1}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2d95246c2816e550018d080c58c60240_l3.png "Rendered by QuickLaTeX.com")
 , we get
, we get ![\[\sum_{i=0}^\infty (1-x)^i = x^{-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-726e8fae76877e0ec1f03efab333ec2e_l3.png "Rendered by QuickLaTeX.com")
 . With a little effort, one can check that the same formula
. With a little effort, one can check that the same formula![\[\sum_{i=0}^\infty (I-X)^i = X^{-1}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1389cddfba9ee14bcbf4293f018c78c9_l3.png "Rendered by QuickLaTeX.com")
 satisfying
satisfying  . These conditions hold for the matrix
. These conditions hold for the matrix  since
since ![\[\left(\frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^{-1} = \sum_{i=0}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8d1be156303c168d2de83dbde76e44d6_l3.png "Rendered by QuickLaTeX.com")
![\[\left(\frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^{-1} = \sum_{i=0}^{t-1} \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i + \sum_{i=t}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-08f4b1a705439315fe0fc038e059d01c_l3.png "Rendered by QuickLaTeX.com")
 , we obtain
, we obtain ![\[\expect[x_t] = x_\star - \left[\sum_{i=t}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i \right]\cdot\frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d973041dba49ba471bbfb6b3d4c2b463_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[x_t] - x_\star = - \left(I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^t \left[\sum_{i=0}^\infty \left( I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^i \right]\cdot\frac{A^\top b}{\norm{A}_{\rm F}^2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8238e65e7b7df2013504f2a6f71799e6_l3.png "Rendered by QuickLaTeX.com")
![\[\expect[x_t] - x_\star = - \left(I - \frac{A^\top A}{\norm{A}_{\rm F}^2}\right)^t x_\star.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3cbe72663f8ef13c762213b5592bb5d2_l3.png "Rendered by QuickLaTeX.com")
![\[\norm{\expect[x_t] - x_\star}^2 \le \norm{I - \frac{A^\top A}{\norm{A}_{\rm F}^2}}^{2t} \norm{x}_\star^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e663370119bd02900da0aa4f7b6faf03_l3.png "Rendered by QuickLaTeX.com")
 . Let
. Let  be a (
be a ( . Then
. Then ![\[I - \frac{A^\top A}{\norm{A}_{\rm F}^2} = I - V \cdot\frac{\Sigma^2}{\norm{A}_{\rm F}^2} \cdot V^\top = V \left( I - \frac{\Sigma^2}{\norm{A}_{\rm F}^2}\right)V^\top.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-468566feb9b39f9e0dd616b47f324ad5_l3.png "Rendered by QuickLaTeX.com")
![\[I - \Sigma^2/\norm{A}_{\rm F}^2 = \diag(1 - \sigma^2_{\rm max}(A)/\norm{A}_{\rm F}^2,\ldots,1-\sigma_{\rm min}^2(A)/\norm{A}_{\rm F}^2).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4bc7dac794d8913157b82fd0df23e41d_l3.png "Rendered by QuickLaTeX.com")
 . We have invoked the definition of the Demmel condition number (2). Therefore, plugging into (7), we obtain
. We have invoked the definition of the Demmel condition number (2). Therefore, plugging into (7), we obtain ![\[\norm{\expect[x_t] - x_\star}^2 \le \left(1-\kappa_{\rm dem}^{-2}\right)^{2t} \norm{x}_\star^2,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7c29ce0386ec23c163afab561f181af1_l3.png "Rendered by QuickLaTeX.com")
 is a matrix and
is a matrix and  is a vector. For simplicity, we assume that this system is
is a vector. For simplicity, we assume that this system is  , randomized Kaczmarz repeatedly performs the following steps:
, randomized Kaczmarz repeatedly performs the following steps: according to the probability distribution
according to the probability distribution  . Here, and going forward,
. Here, and going forward,  denotes the
denotes the  is the
is the  .
. and
and  are not very large, this is an easy problem: Simply sweep through all the rows
are not very large, this is an easy problem: Simply sweep through all the rows  and compute their norms
and compute their norms  . Then, sampling can be done using any algorithm for sampling from a weighted list of items. But what if
. Then, sampling can be done using any algorithm for sampling from a weighted list of items. But what if ![\[\norm{a_i}^2 \le B \quad \text{for each } i = 1,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a80c9da38c2d98b188cb70f64c8637e8_l3.png "Rendered by QuickLaTeX.com")
![[-1,1]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-634fe4705a526f221e312cffd3ceda93_l3.png "Rendered by QuickLaTeX.com") , then (1) holds with
, then (1) holds with  . To sample a row
. To sample a row  , we perform the following the rejection sampling procedure:
, we perform the following the rejection sampling procedure:
 uniformly at random (i.e.,
uniformly at random (i.e.,  is equally likely to be any row index between
is equally likely to be any row index between  , accept and set
, accept and set  . With the remaining probability
. With the remaining probability  , reject and return to step 1.
, reject and return to step 1. . Our goal is to choose a random index
. Our goal is to choose a random index  , i.e.,
, i.e.,  . We will call
. We will call  the target distribution. (Note that we do not require the weights
the target distribution. (Note that we do not require the weights  .)
.) , i.e., we can efficiently generate random
, i.e., we can efficiently generate random  . Further, suppose that the proposal distribution dominates the target distribution in the sense that
. Further, suppose that the proposal distribution dominates the target distribution in the sense that ![\[w_i \le \rho_i \quad \text{for each } i=1,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7f2901cd8e565853465062e41a59b5bd_l3.png "Rendered by QuickLaTeX.com")
 , accept and set
, accept and set  and
and  for all
for all  , after which the probability of accepting
, after which the probability of accepting  . Therefore, the probability of outputting
. Therefore, the probability of outputting ![\[\prob \{\text{$i$ accepted this loop}\} = \frac{\rho_i}{\sum_{j=1}^n \rho_j} \cdot \frac{w_i}{\rho_i} = \frac{w_i}{\sum_{j=1}^n \rho_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8fb5712ddc38527600d4fd51394bfc7_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{\text{any accepted this loop}\} = \sum_{i=1}^n \prob \{\text{$i$ accepted this loop}\} = \frac{\sum_{i=1}^n w_i}{\sum_{i=1}^n \rho_i}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-87004ae4b99269f6917de03d3a4ec86e_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{\text{$i$ accepted this loop} \mid \text{any accepted this loop}\} = \frac{\prob \{\text{$i$ accepted this loop}\}}{\prob \{\text{any accepted this loop}\}} = \frac{w_i}{\sum_{j=1}^n w_j}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8740003cf2cc5dfd18def3387663bcc2_l3.png "Rendered by QuickLaTeX.com")
 , the ratio of the total mass of the target
, the ratio of the total mass of the target  . Thus, rejection sampling will have a high acceptance rate if
. Thus, rejection sampling will have a high acceptance rate if  and a low acceptance rate if
and a low acceptance rate if  . The conclusion for practice is that rejection sampling is only computationally efficient if one has access to a good proposal distribution
. The conclusion for practice is that rejection sampling is only computationally efficient if one has access to a good proposal distribution  .
. , but this not all that rejection sampling can do. Indeed, one can also use rejection sampling for sampling a real-valued parameters
, but this not all that rejection sampling can do. Indeed, one can also use rejection sampling for sampling a real-valued parameters  or a multivariate parameter
or a multivariate parameter  from a given (unnormalized) probability density function (or, if one likes, an unnormalized probability measure).
from a given (unnormalized) probability density function (or, if one likes, an unnormalized probability measure). and
and  are necessary to define the sampling probabilities
are necessary to define the sampling probabilities  and
and  , as computing
, as computing  requires a full pass over the data to compute.
requires a full pass over the data to compute. denote the initial matrix and
denote the initial matrix and  denote a trivial initial approximation. For
denote a trivial initial approximation. For  , RPCholesky performs the following steps:
, RPCholesky performs the following steps: with probability
with probability  .
. . Here,
. Here,  denotes the
denotes the  .
. .
. . With an optimized implementation, RPCholesky requires only
. With an optimized implementation, RPCholesky requires only  operations and evaluates just
operations and evaluates just  entries of the input matrix
entries of the input matrix 
 denote the first
denote the first  produced by
produced by ![\[\hat{A}^{(j)} = A(:,S_j) A(S_j,S_j)^{-1}A(S_j,:). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ebc63948bd7d20eda4ccafa74555ba6b_l3.png "Rendered by QuickLaTeX.com")
![\[A^{(j)} = A - \hat{A}^{(j)} = A - A(:,S_j) A(S_j,S_j)^{-1}A(S_j,:).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc5136dd307c099cb5fa70663104166b_l3.png "Rendered by QuickLaTeX.com")
![\[A^{(j)}_{ii} = A_{ii} - A(i,S_j) A(S_j,S_j)^{-1} A(S_j,i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8442521e6a6003a935f35d25aca1c73_l3.png "Rendered by QuickLaTeX.com")
 is cheap, just requiring some arithmetic involving matrices and vectors of size
is cheap, just requiring some arithmetic involving matrices and vectors of size  , but it is expensive to evaluate all of the
, but it is expensive to evaluate all of the  . (One may verify that, as required,
. (One may verify that, as required,  for all
for all  , sample random indices
, sample random indices  using rejection sampling with proposal distribution
using rejection sampling with proposal distribution  are evaluated on an as-needed basis using (2).
are evaluated on an as-needed basis using (2). be a domain and let
be a domain and let  be a (
be a ( . We consider the task of evaluating
. We consider the task of evaluating ![\[I[f] = \int_\Omega f(x) g(x) \, \mathrm{d}\mu(x).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4767401252e7558c87b5a518f3609468_l3.png "Rendered by QuickLaTeX.com")
 ,
, ![I[f]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-603f22dedcf932b6f47e37160c09afd8_l3.png "Rendered by QuickLaTeX.com") for multiple different functions
for multiple different functions ![\[\hat{I}_{w,s}[f] = \sum_{i=1}^n w_i f(s_i) \approx I[f].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4bb6a83827b630719dd50338a790b9c6_l3.png "Rendered by QuickLaTeX.com")
 and points
and points  such that the approximation
such that the approximation ![\hat{I}_{w,s}[f] \approx I[f]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-61580a8cc2043483ce9c1ab09e3ea85e_l3.png "Rendered by QuickLaTeX.com") is accurate.
is accurate.
 be an RKHS with
be an RKHS with  . We can interpret the norm as assigning a roughness
. We can interpret the norm as assigning a roughness  to each function
to each function  . It is related to the RKHS
. It is related to the RKHS  by the reproducing property
by the reproducing property![\[f(x)=\langle f, k(x,\cdot)\rangle \quad \text{for every }f\in\mathcal{H},x\in\Omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-276db3513a1ea34da49f342ad7f4a7ea_l3.png "Rendered by QuickLaTeX.com")
 represents the univariate function obtained by setting the first input of
represents the univariate function obtained by setting the first input of  . Let’s first assume that the nodes
. Let’s first assume that the nodes  that we’ll called the ideal weights. There (at least) are five equivalent ways of characterizing the ideal weights. We’ll present all of them. As an exercise, you can try and convince yourself that these characterizations are equivalent, giving rise to the same weights.
that we’ll called the ideal weights. There (at least) are five equivalent ways of characterizing the ideal weights. We’ll present all of them. As an exercise, you can try and convince yourself that these characterizations are equivalent, giving rise to the same weights. .
.![\[\hat{I}_{w_\star,s}[k(s_i,\cdot)]=I[k(s_i,\cdot)] \quad \text{for } i=1,2,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-52bf51e5f4ddb42195d5896ff71ebe02_l3.png "Rendered by QuickLaTeX.com")
 :
:![\[\sum_{j=1}^n k(s_i,s_j)w^\star_j = \int_\Omega k(s_i,x) g(x)\,\mathrm{d}\mu(x) \quad \text{for }i=1,2,\ldots,n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1a5ca36966f56dae9d994e208f9ef036_l3.png "Rendered by QuickLaTeX.com")
 is the ideal weights.
is the ideal weights.
 at the nodes, obtaining an interpolant
at the nodes, obtaining an interpolant  . Then, obtain an approximation to the integral by integrating the interpolant:
. Then, obtain an approximation to the integral by integrating the interpolant:![\[\hat{I}_{w^\star,s}[f] \coloneqq \int_\Omega \hat{f}(x) g(x) \, \mathrm{d}\mu(x).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0c68060fdf085f41d9cbdd9cc5c16e31_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{f} = \argmin \{ \norm{h} : h(s_i) = f(s_i) \text{ for } i=1,\ldots,n\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5619a986180c554d2a079e5156a97571_l3.png "Rendered by QuickLaTeX.com")
![\[\hat{f} = \sum_{i=1}^n \alpha_i k(\cdot,s_i)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0abcded0eda92deac99b904817afdf5b_l3.png "Rendered by QuickLaTeX.com")
 .With a little algebra, you can show that the integral of
.With a little algebra, you can show that the integral of ![\[I[\hat{f}] = \sum_{i=1}^n w^\star_i f(s_i),\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f0da23bc7dc97894b0a6702b6bf6ddfb_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{Err}(w,s)=\sup_{\norm{f}\le 1}\left| I[f] - \hat{I}_{w,s}[f]\right|.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6e46307dc4cdac54a3ab010b36ed7858_l3.png "Rendered by QuickLaTeX.com")
 is the highest possible quadrature error for a function
is the highest possible quadrature error for a function  of norm at most 1.
of norm at most 1.
![\[w^\star=\operatorname*{argmin}_{w\in\real^n}\operatorname{Err}(w,s).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06e68886b8279d9d592cae3775d4295f_l3.png "Rendered by QuickLaTeX.com")
 for a mean-zero Gaussian process with
for a mean-zero Gaussian process with ![\[\Cov(f(x),f(y))=k(x,y)\quad \text{for every } x,y\in\Omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-19fea8ea8406c891c0d6bab2166dc4a4_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{MSE}(w,s)\coloneqq \expect_{f\sim\operatorname{GP}(0,k)} \left( I[f] - \hat{I}_{w,s}[f] \right)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4741e0bcba7276b0a3a9266cd61b54c0_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{MSE}(w,s)=\operatorname{Err}(w,s)^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc387f1211b9e5a070f2778a8205c022_l3.png "Rendered by QuickLaTeX.com")
![\[w^\star=\operatorname*{argmin}_{w\in\real^n}\operatorname{MSE}(w,s).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-32506adbc7176da52a920dbf4c373149_l3.png "Rendered by QuickLaTeX.com")
 . The integral of this random function
. The integral of this random function ![I[h]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e47eba0764d69669e85367cf245d71d3_l3.png "Rendered by QuickLaTeX.com") is a random variable. To numerically integrate a function
is a random variable. To numerically integrate a function  agreeing with
agreeing with ![\[\hat{I}_{w^\star,s}[f]\coloneqq \expect_{h\sim\operatorname{GP}(0,k)}[I[h] \mid h(s_i)=f(s_i) \text{ for } i=1,\ldots,n].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ce1156fb9da612607ae0158770a34645_l3.png "Rendered by QuickLaTeX.com")
 , it is reasonable to add an additional constraint that the weights
, it is reasonable to add an additional constraint that the weights ![\[w\in\Delta\coloneqq \left\{ p\in\real^n_+ : \sum_{i=1}^n p_i = 1\right\}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-55c593b2593abc9ea8c18d4f8edbccd3_l3.png "Rendered by QuickLaTeX.com")
 ; thus, in effect, quadrature amounts to approximating one probability measure
; thus, in effect, quadrature amounts to approximating one probability measure  . Additional constraints such as these can easily be imposed when using the optimization characterizations 3 and 4 of the ideal weights. See
. Additional constraints such as these can easily be imposed when using the optimization characterizations 3 and 4 of the ideal weights. See  ? To pick the nodes, it seems sensible to try and minimize the worst-case error
? To pick the nodes, it seems sensible to try and minimize the worst-case error  with the ideal weights
with the ideal weights ![\[\operatorname{Err}(w^\star,s) = \norm{\int_\Omega (k(\cdot,x) - \hat{k}_s(\cdot,x)) g(x) \, \mathrm{d}\mu(x)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f66a0cdfc8a943a944dad0fed98dbad9_l3.png "Rendered by QuickLaTeX.com")
 is the
is the ![\[\hat{k}_s(x,y) = k(x,s) k(s,s)^{-1} k(s,y).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9c55d13be5a7cdb27139303cb08ec19f_l3.png "Rendered by QuickLaTeX.com")
 for the kernel matrix with
for the kernel matrix with  entry
entry  and
and  and
and  for the row and column vectors with
for the row and column vectors with  and
and  .
.
 as accurate as possible.
as accurate as possible. is and, thus, the smaller the error
is and, thus, the smaller the error  , it has a high probability of placing the next node
, it has a high probability of placing the next node  far from the previously selected nodes.
far from the previously selected nodes.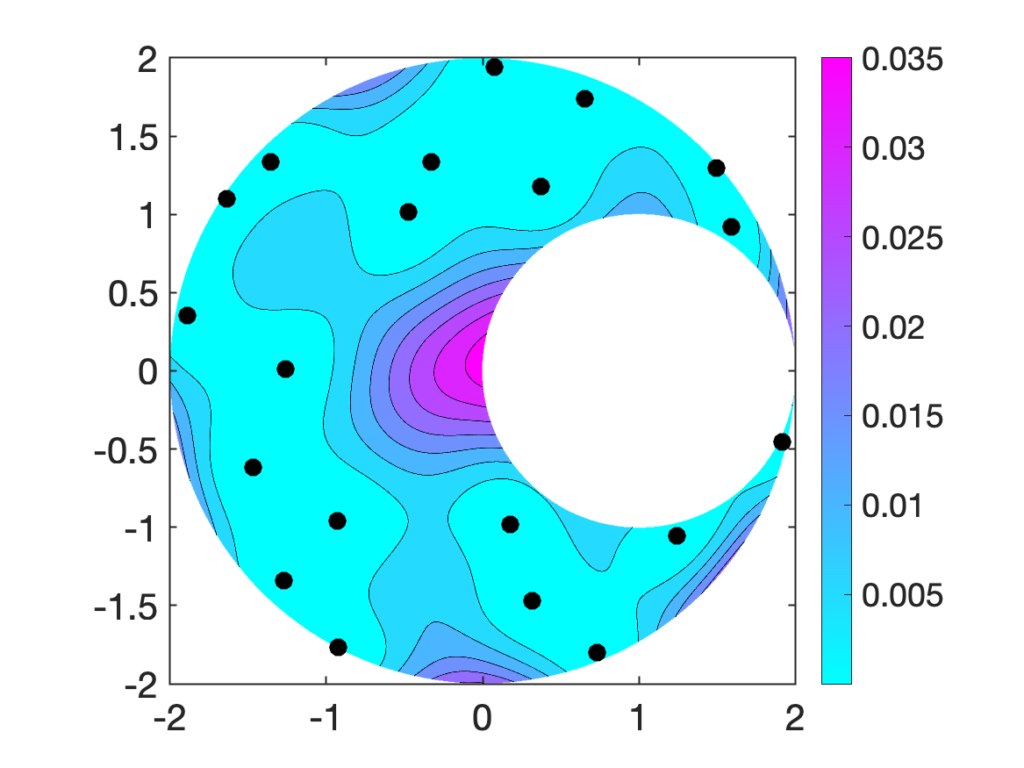
 seeks to compress a high-dimensional matrix
seeks to compress a high-dimensional matrix  or vector
or vector  to a lower-dimensional sketched matrix
to a lower-dimensional sketched matrix  or vector
or vector  . The quality of a sketching matrix for a matrix
. The quality of a sketching matrix for a matrix  , defined to be the smallest number
, defined to be the smallest number  such that
such that ![\[(1-\varepsilon) \norm{x} \le \norm{Sx} \le (1+\varepsilon) \norm{x} \quad \text{for every } x \in \operatorname{col}(A).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06811a2d05bdccddd7b3c3079ed47729_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  .
. matrix.
matrix. (one million) and output dimension
(one million) and output dimension  . For the SRTT, we use the
. For the SRTT, we use the  .
.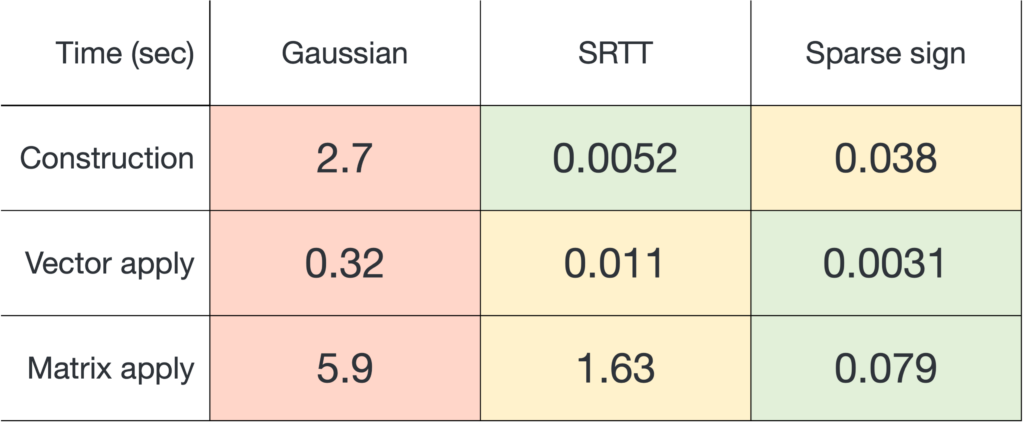
 and applying it to a matrix
and applying it to a matrix  , sparse sign embeddings are 14× faster than SRTTs and 73× faster than Gaussian embeddings.
, sparse sign embeddings are 14× faster than SRTTs and 73× faster than Gaussian embeddings. for
for  and
and  using the MATLAB
using the MATLAB  (equivalently,
(equivalently,  ) is shown as a dashed line.
) is shown as a dashed line.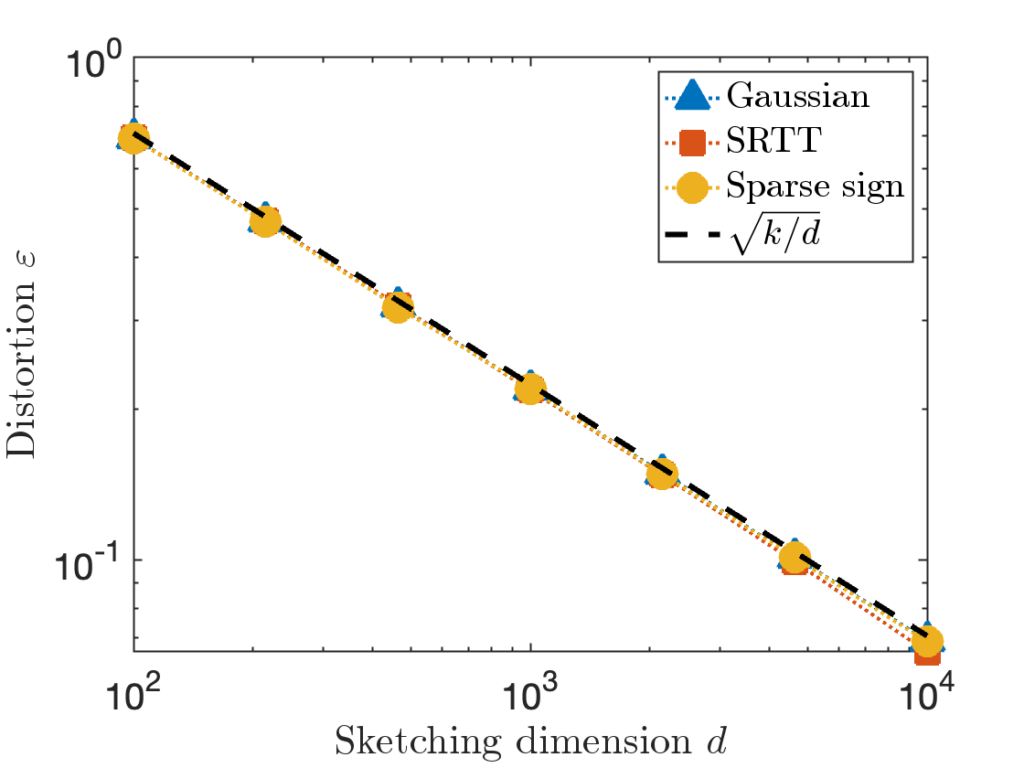
 is taken to be a matrix with independent standard Gaussian random values.
is taken to be a matrix with independent standard Gaussian random values. is taken to be the
is taken to be the  is taken to be the
is taken to be the  identity matrix stacked onto a
identity matrix stacked onto a  matrix of zeros.
matrix of zeros.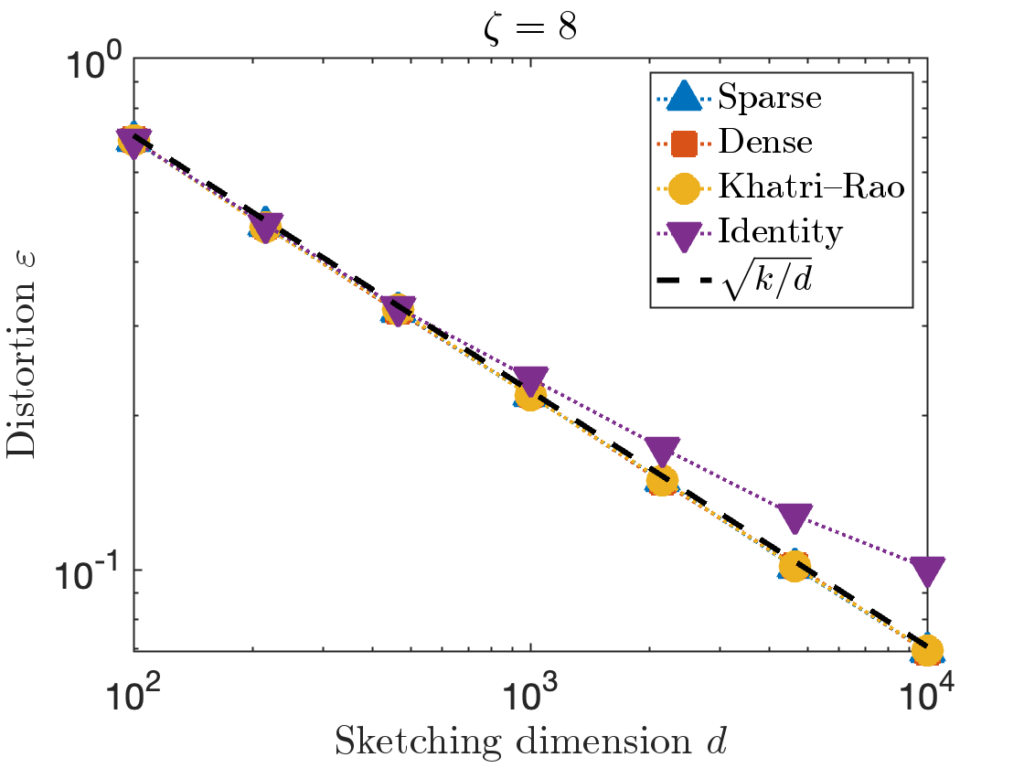
 . However, for the last test matrix “Identity”, we see the distortion begins to slightly exceed this predicted distortion for
. However, for the last test matrix “Identity”, we see the distortion begins to slightly exceed this predicted distortion for  .
. , we can increase the value of the sparsity parameter
, we can increase the value of the sparsity parameter  . We recommend
. We recommend ![\[\zeta = \max \left( 8 , \left\lceil 2\sqrt{\frac{d}{k}} \right\rceil \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5f1ebe7695d9918399b0f97b628fcce1_l3.png "Rendered by QuickLaTeX.com")
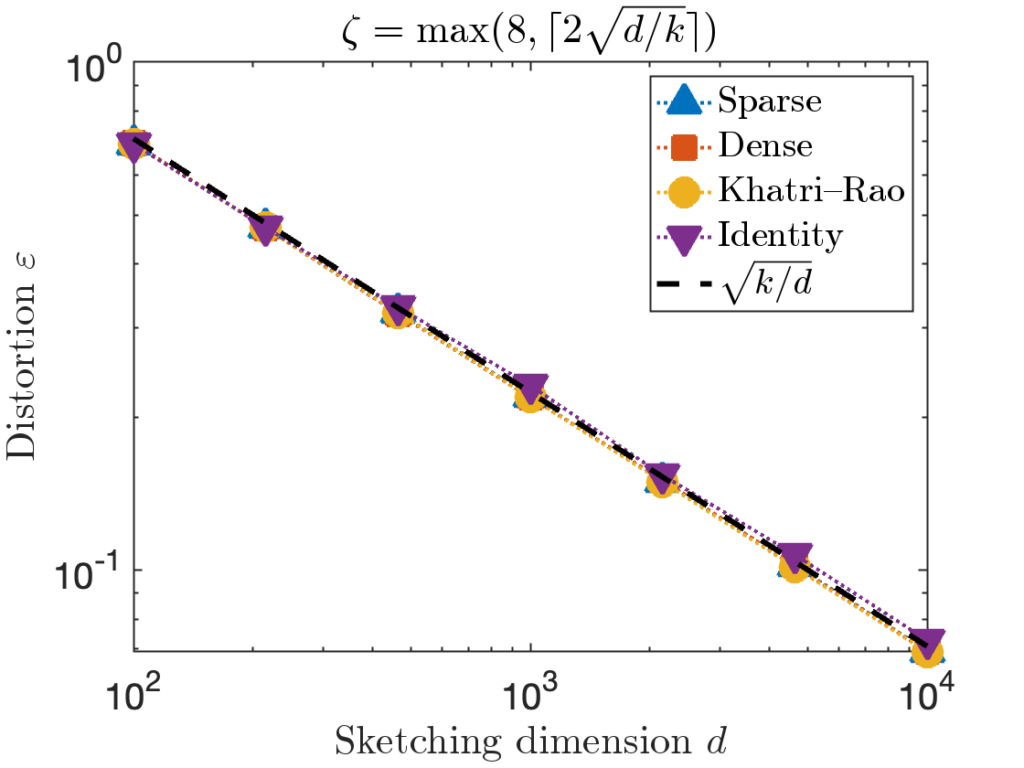
 can be necessary to achieve the optimal distortion.
can be necessary to achieve the optimal distortion.![\[S = \frac{1}{\sqrt{\zeta}} \begin{bmatrix} s_1 & \cdots & s_n \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4871e53b2bb59ad11946f2d1eafc66f7_l3.png "Rendered by QuickLaTeX.com")
 is an independent and randomly generated to contain exactly
is an independent and randomly generated to contain exactly  or
or  with 50/50 odds.
with 50/50 odds.
![\[(1-\varepsilon) \norm{x} \le \norm{Sx} \le (1+\varepsilon) \norm{x} \quad \text{for all }x \in \operatorname{col}(A).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-41fd14292c1c85cd318dde801a75e1bd_l3.png "Rendered by QuickLaTeX.com")
![\[d = \frac{k}{\varepsilon^2} \quad \text{and} \quad \zeta = \max\left(8,\frac{2}{\varepsilon}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ed85cec8dadffec7c7a3b4e099d6637d_l3.png "Rendered by QuickLaTeX.com")
 . The value
. The value  has demonstrated deficiencies and should almost always be avoided (see below). The scaling
has demonstrated deficiencies and should almost always be avoided (see below). The scaling  is derived from the
is derived from the ![\[d = \mathcal{O} \left( \frac{k \log k}{\varepsilon^2} \right) \quad \text{and} \quad \zeta = \mathcal{O}\left( \frac{\log k}{\varepsilon} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4fb6d0d60c85bc8a60d95bb0a19e63d6_l3.png "Rendered by QuickLaTeX.com")
 factor and the lack of explicit constants in the
factor and the lack of explicit constants in the  and can also be generated using a single line. The real challenge to generating sparse sign embeddings in MATLAB is the row indices, since each batch of
and can also be generated using a single line. The real challenge to generating sparse sign embeddings in MATLAB is the row indices, since each batch of  sparse sign embedding with sparsity
sparse sign embedding with sparsity  and weights
and weights  . To apply
. To apply  and reweight them using the weights:
and reweight them using the weights:![\[b \in \real^n \longmapsto Sb = (w_1 b_{i_1},\ldots,w_db_{i_d}) \in \real^d.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-712b97fc6f6efb9bf9a40dac2a652f5c_l3.png "Rendered by QuickLaTeX.com")
 for the hard “Identity” test matrix used above.
for the hard “Identity” test matrix used above.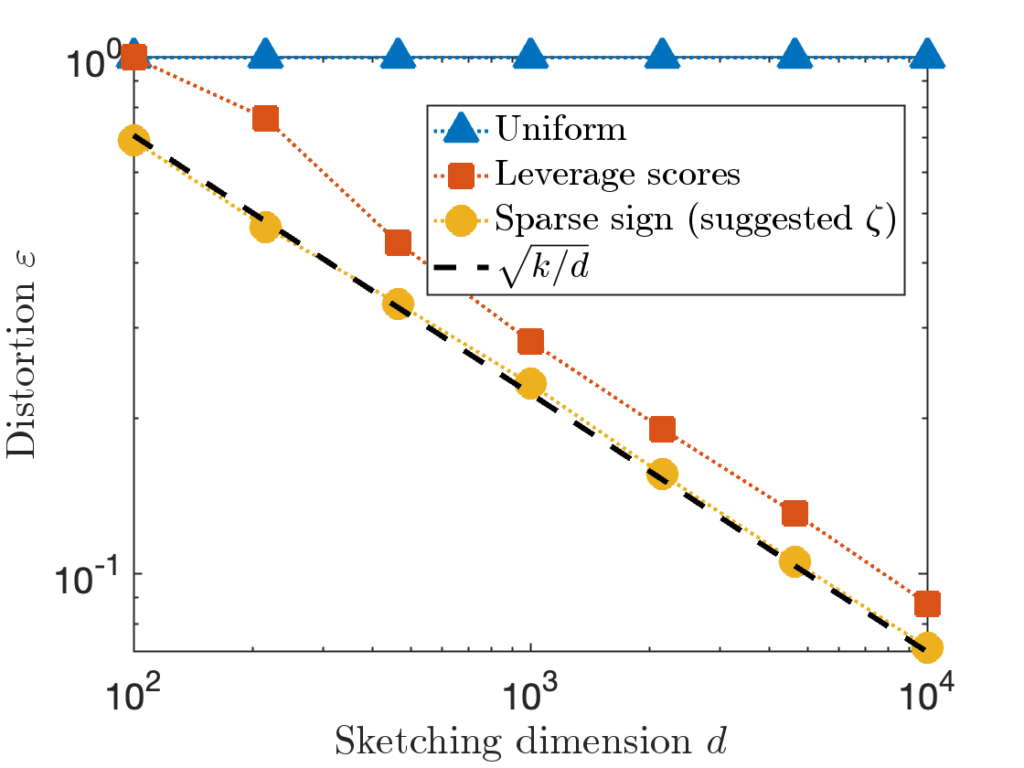
 cost, much higher than other types of sketches.
cost, much higher than other types of sketches. or row of the matrix
or row of the matrix  , and compare the distortion of CountSketch to the sparse sign embedding with parameters
, and compare the distortion of CountSketch to the sparse sign embedding with parameters  :
: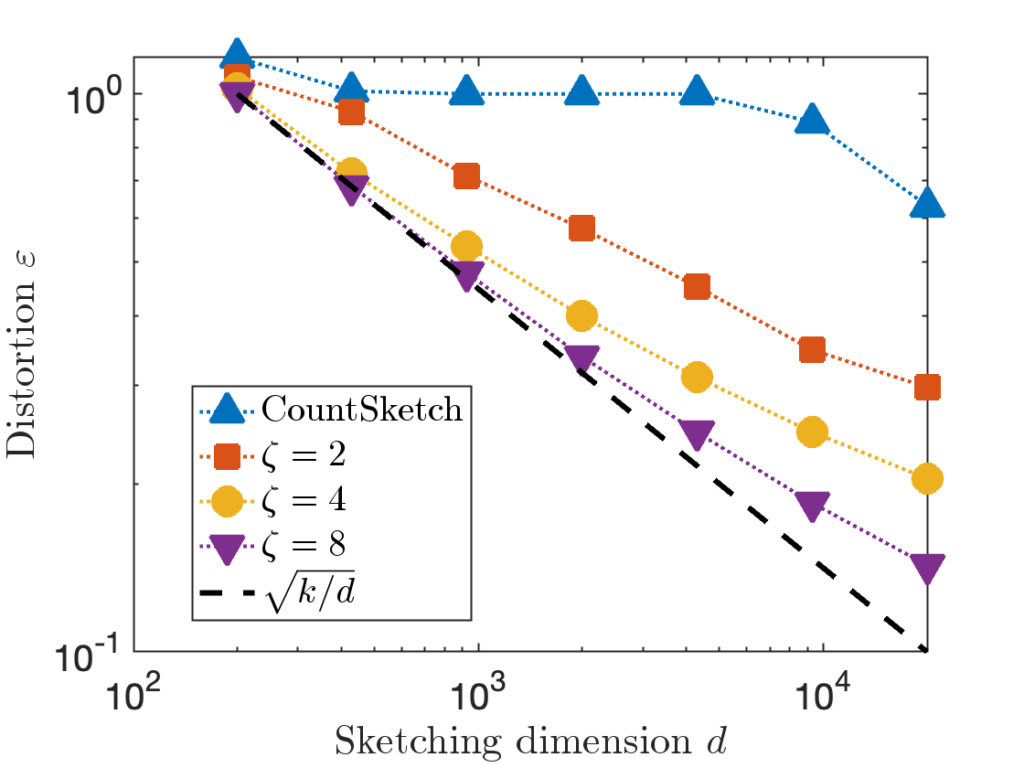
 , 20× higher than
, 20× higher than  in order to achieve distortion
in order to achieve distortion  (or perhaps
(or perhaps  ). This difference between
). This difference between  for CountSketch and
for CountSketch and  for other sketching matrices is a at the root of CountSketch’s woefully bad performance on some inputs.
for other sketching matrices is a at the root of CountSketch’s woefully bad performance on some inputs. is an informal symbol meaning “proportional to”.
is an informal symbol meaning “proportional to”. . This is Theorem 16 in
. This is Theorem 16 in  , where
, where  is a CountSketch of size
is a CountSketch of size  and
and  is a Gaussian sketching matrix of size
is a Gaussian sketching matrix of size  .
. is a CountSketch matrix with output dimension
is a CountSketch matrix with output dimension  , then the distortion of
, then the distortion of  with high probability.
with high probability.![\[SA = \begin{bmatrix} s_1 & \cdots & s_k \end{bmatrix}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-82c7ea0e5c52b6bd9bdb389f3d3f92d7_l3.png "Rendered by QuickLaTeX.com")
 has a single
has a single  in a uniformly random location
in a uniformly random location  .
.
 are not all different from each other, say
are not all different from each other, say  . Set
. Set  , where
, where  is the standard basis vector with
is the standard basis vector with  but
but  . Thus, for the distortion relation
. Thus, for the distortion relation ![\[(1-\varepsilon) \norm{x} =(1-\varepsilon)\sqrt{2} \le 0 = \norm{(SA)x}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b08feebb30829a0e73b02188eba6f901_l3.png "Rendered by QuickLaTeX.com")
 . Thus,
. Thus, ![\[\prob \{ \varepsilon \ge 1 \} \ge \prob \{ j_1,\ldots,j_k \text{ are not distinct} \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-12fa7b716b97c7e9482f4f6327bd4728_l3.png "Rendered by QuickLaTeX.com")
 pairs of people. Each pair of people has a
pairs of people. Each pair of people has a  chance of sharing a birthday, so the expected number of birthdays in a room of 23 people is
chance of sharing a birthday, so the expected number of birthdays in a room of 23 people is  . Since are 0.69 birthdays shared on average in a room of 23 people, it is perhaps less surprising that 23 is the critical number at which the chance of two people sharing a birthday exceeds 50%.
. Since are 0.69 birthdays shared on average in a room of 23 people, it is perhaps less surprising that 23 is the critical number at which the chance of two people sharing a birthday exceeds 50%. and
and  in CountSketch have a
in CountSketch have a  chance of being the same. There are
chance of being the same. There are  pairs of indices, so the expected number of equal indices
pairs of indices, so the expected number of equal indices  . Thus, we should anticipate
. Thus, we should anticipate  is required to ensure that
is required to ensure that ![\[\prob \{ j_1,\ldots,j_k \text{ are not distinct} \} = 1 - \prob \{ j_1,\ldots,j_k \text{ are distinct} \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-338a38501a565185b794f8ed03803c9a_l3.png "Rendered by QuickLaTeX.com")
 are all distinct, the probability
are all distinct, the probability  are distinct is just the probability that
are distinct is just the probability that  values
values ![\[\prob\{ j_1,\ldots,j_i \text{ are distinct} \mid j_1,\ldots,j_{i-1} \text{ are distinct}\} = 1 - \frac{i-1}{d}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b474f00c44e53b7c3c398d56121e1ac3_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{ j_1,\ldots,j_k \text{ are distinct} \} = \prod_{i=1}^k \left(1 - \frac{i-1}{d} \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7dd28281f8dbed8b3a1452a21e7410c8_l3.png "Rendered by QuickLaTeX.com")
 for every
for every ![\[\mathbb{P} \{ j_1,\ldots,j_k \text{ are distinct} \} \le \prod_{i=0}^{k-1} \exp\left(-\frac{i}{d}\right) = \exp \left( -\frac{1}{d}\sum_{i=0}^{k-1} i \right) = \exp\left(-\frac{k(k-1)}{2d}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8d9d4c7d12af0fd89e53ca261c5df287_l3.png "Rendered by QuickLaTeX.com")
![\[\prob \{ \varepsilon \ge 1 \} \ge 1-\prob \{ j_1,\ldots,j_k \text{ are distinct} \\}\ge 1-\exp\left(-\frac{k(k-1)}{2d}\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-144cfc73d778c4f9ed7442f5a6610e5e_l3.png "Rendered by QuickLaTeX.com")
![\[\prob\{\varepsilon \ge 1\} \ge \frac{1}{2} \quad \text{if} \quad d \le \frac{k(k-1)}{2\ln 2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e11567078a415b79b06a8e5e6f0b1600_l3.png "Rendered by QuickLaTeX.com")
 . A sketching matrix is a
. A sketching matrix is a  matrix
matrix  . When multiplied into a high-dimensional vector
. When multiplied into a high-dimensional vector 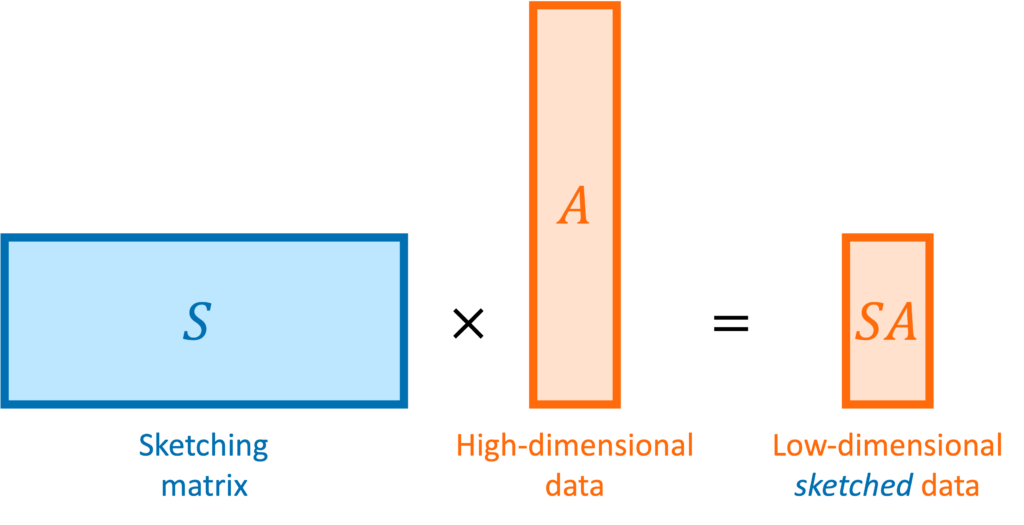
 be a collection of vectors. For
be a collection of vectors. For  , we require that
, we require that  :
: ![\[(1-\varepsilon) \norm{x}\le\norm{Sx}\le(1+\varepsilon)\norm{x} \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-735df49a45ff5d0bb27d478569004bdd_l3.png "Rendered by QuickLaTeX.com")
![\[\operatorname{col}(A) \coloneqq \{ Ax : x \in \real^k \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5ecde6b43df29caba14fd43682b4cc86_l3.png "Rendered by QuickLaTeX.com")
 with an output dimension of roughly
with an output dimension of roughly  . In particular, the sketching dimension
. In particular, the sketching dimension  in the column space of
in the column space of  elements) or dimension (
elements) or dimension ( requires roughly
requires roughly  operations, rather than the
operations, rather than the  operations we would expect to multiply a
operations we would expect to multiply a  is
is ![\[S = \sqrt{\frac{n}{d}} \cdot R \cdot F \cdot D.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-92095cbf9c7d55273bd572a33debe415_l3.png "Rendered by QuickLaTeX.com")
 is a diagonal matrix whose entries are each a random
is a diagonal matrix whose entries are each a random  (chosen independently with equal probability).
(chosen independently with equal probability). is a fast trigonometric transform such as a fast
is a fast trigonometric transform such as a fast  is a selection matrix. To generate
is a selection matrix. To generate  , let
, let  , selected without replacement.
, selected without replacement.  for every vector
for every vector  , and
, and  operations, a significant improvement over the
operations, a significant improvement over the  , larger than for a Gaussian sketch.
, larger than for a Gaussian sketch.![\[S = \frac{1}{\sqrt{\zeta}} \begin{bmatrix} s_1 & s_2 & \cdots & s_n \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bb5806806e2e5900d4351c08befbd82e_l3.png "Rendered by QuickLaTeX.com")
 nonzero entries. The parameter
nonzero entries. The parameter  in practice.
in practice. or
or  operations) to apply to a vector, depending on parameter choices (see below). With a good sparse matrix library, sparse sign embeddings are often the fastest sketching matrix by a wide margin.
operations) to apply to a vector, depending on parameter choices (see below). With a good sparse matrix library, sparse sign embeddings are often the fastest sketching matrix by a wide margin. random numbers, higher than SRTTs (roughly
random numbers, higher than SRTTs (roughly  numbers).
numbers).  ; the theoretically sanctioned sketching dimension (at least according to existing theory) is larger than for a Gaussian sketch. In practice, we can often get away with using
; the theoretically sanctioned sketching dimension (at least according to existing theory) is larger than for a Gaussian sketch. In practice, we can often get away with using  .
. operations. Therefore, sketching offers the promise of speeding up linear algebraic computations involving
operations. Therefore, sketching offers the promise of speeding up linear algebraic computations involving ![\[\operatorname*{minimize}_{x\in\real^k} \norm{Ax - b}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-07528304ce875f54aa1173062c5ec10a_l3.png "Rendered by QuickLaTeX.com")
 .
.
 . Applying
. Applying ![\[\operatorname*{minimize}_{\hat{x}\in\real^k} \norm{(SA)\hat{x} - Sb}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7110d3e9c1dd7e29793603a5bafdd201_l3.png "Rendered by QuickLaTeX.com")
 is the sketch-and-solve solution to the least-squares problem, which we can use as an approximate solution to the original least-squares problem.
is the sketch-and-solve solution to the least-squares problem, which we can use as an approximate solution to the original least-squares problem.
 , first apply sketching to obtain
, first apply sketching to obtain  and then apply an out-of-the-box clustering algorithms like
and then apply an out-of-the-box clustering algorithms like ![\[\norm{A\hat{x} - b} \le \frac{1+\varepsilon}{1-\varepsilon} \norm{Ax - b}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3d1d034bd8bf93f1c15e435a27a355ee_l3.png "Rendered by QuickLaTeX.com")
 , then this bound tells us that
, then this bound tells us that ![\[\norm{A\hat{x} - b} \le 2\norm{Ax_\star - b}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-588170743415f491f48ef2adc3d46628_l3.png "Rendered by QuickLaTeX.com")
 . For such applications, the bound (4) ensures that
. For such applications, the bound (4) ensures that  . Often, this means
. Often, this means  , measuring how close
, measuring how close  and residual norm
and residual norm  ). Then, we generate a sparse sign embedding of dimension
). Then, we generate a sparse sign embedding of dimension  ). Then, we compute the sketch-and-solve solution and, as reference, a “direct” solution by
). Then, we compute the sketch-and-solve solution and, as reference, a “direct” solution by  , close to direct method’s residual norm of
, close to direct method’s residual norm of  . However, the forward error of sketch-and-solve is
. However, the forward error of sketch-and-solve is  nine orders of magnitude larger than the direct method’s forward error of
nine orders of magnitude larger than the direct method’s forward error of  .
. , to decrease the distortion by a factor of ten requires increasing the sketching dimension
, to decrease the distortion by a factor of ten requires increasing the sketching dimension  that we hope will converge at a rapid rate to the true least-squares solution,
that we hope will converge at a rapid rate to the true least-squares solution,  , then
, then  .
.![\[SA = QR,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9edf84077821e9e0cd0c772b77024799_l3.png "Rendered by QuickLaTeX.com")
![\[A^\top A \approx (SA)^\top (SA) = R^\top Q^\top Q R = R^\top R.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-90e14ee1172525fd795980cb3ef1a647_l3.png "Rendered by QuickLaTeX.com")
 since
since  has orthonormal columns. The conclusion is that
has orthonormal columns. The conclusion is that  .
.
![\[(A^\top A)x = A^\top b. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3f3c5265345a169d6633a759854510da_l3.png "Rendered by QuickLaTeX.com")
 by
by  in (5) and solving. The resulting solution is
in (5) and solving. The resulting solution is![\[x_0 = R^{-1} (R^{-\top}(A^\top b)).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6b63093ea5c1d9714fb0deab43fc4dc3_l3.png "Rendered by QuickLaTeX.com")
 will typically not be a good solution to the least-squares problem (2), so we need to iterate. To do so, we’ll try and solve for the error
will typically not be a good solution to the least-squares problem (2), so we need to iterate. To do so, we’ll try and solve for the error  . To derive an equation for the error, subtract
. To derive an equation for the error, subtract  from both sides of the normal equations (5), yielding
from both sides of the normal equations (5), yielding ![\[(A^\top A)(x-x_0) = A^\top (b-Ax_0).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4f1ee993844dbcd8e397f27ccd1deefb_l3.png "Rendered by QuickLaTeX.com")
 :
: ![\[x\approx x_1 \coloneqq x_0 + R^{-\top}(R^{-1}(A^\top(b-Ax_0))).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0496d7e3c8bc117e658c57fc167081be_l3.png "Rendered by QuickLaTeX.com")
 , approximate
, approximate  , and obtain a new approximate solution
, and obtain a new approximate solution  . Continuing this process, we obtain an iteration
. Continuing this process, we obtain an iteration![\[x_{i+1} = x_i + R^{-\top}(R^{-1}(A^\top(b-Ax_i))).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f8db238fc978e6a8fc968fc5e387ec27_l3.png "Rendered by QuickLaTeX.com")
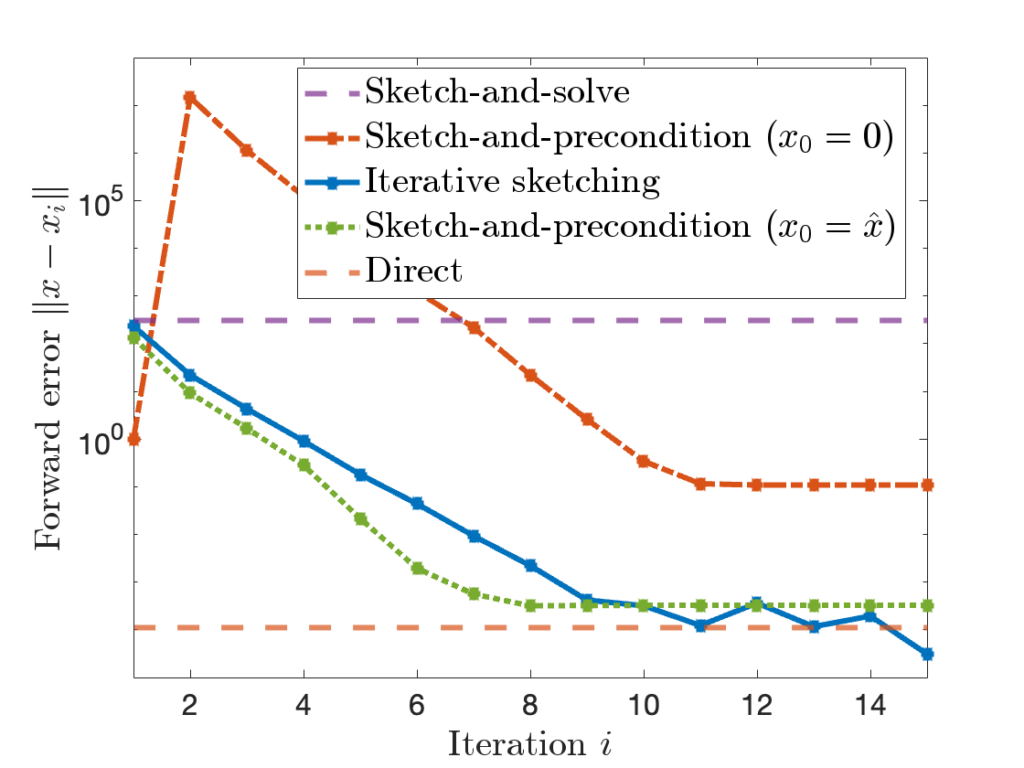
 at every iteration. Later,
at every iteration. Later, 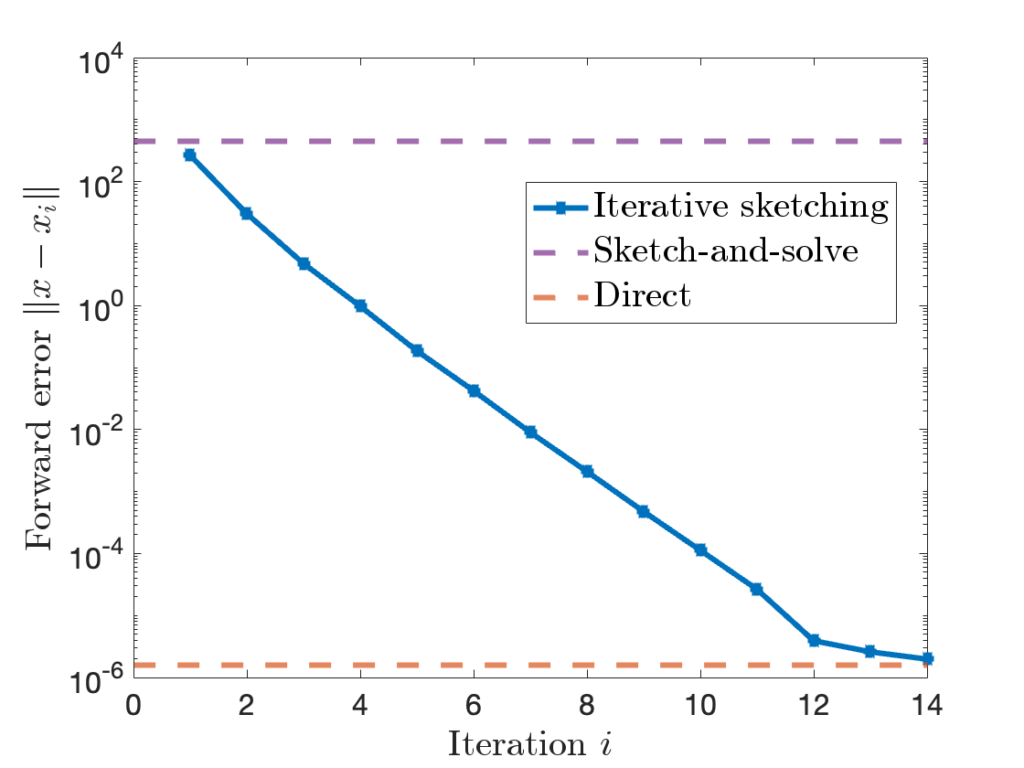
 to
to  by the iteration (6). If we run for enough iterations
by the iteration (6). If we run for enough iterations  can be quite high.
can be quite high.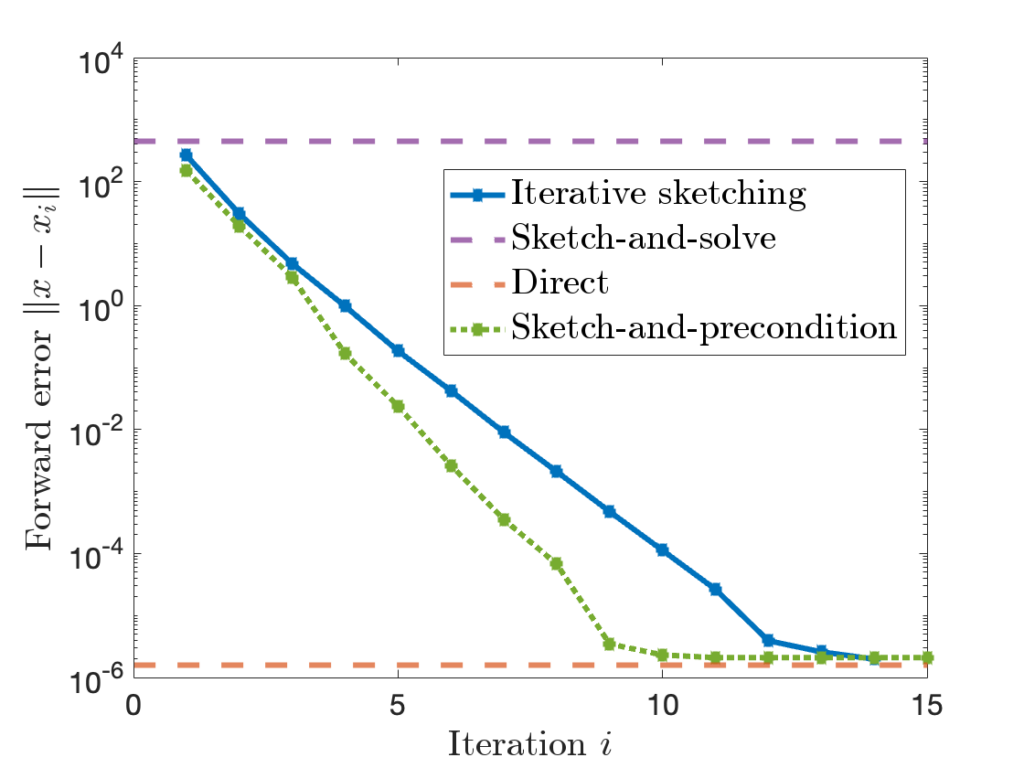
 is comparable to accuracy of a standard
is comparable to accuracy of a standard  for sketch-and-precondition, then sketch-and-precondition appears to be forward stable in practice. No theoretical analysis supporting this finding is known at present.
for sketch-and-precondition, then sketch-and-precondition appears to be forward stable in practice. No theoretical analysis supporting this finding is known at present. and residual
and residual  .
.
 , estimate its trace
, estimate its trace  .
.![\[\hat{\tr} = \frac{1}{m} \sum_{i=1}^m \omega_i^* A \omega_i. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a287886ee24c9ab99dfe35b6cdae83cf_l3.png "Rendered by QuickLaTeX.com")
 are random vectors, usually chosen to be
are random vectors, usually chosen to be  denotes the
denotes the  .
.![\[\mathbb{E} [\omega_i\omega_i^*] = I, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-74bb76367ad1408bd538d049774cf990_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} [\hat{\tr}] = \tr A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f6407c7a741003cc20780e72d050cb56_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} [\hat{\tr}] = \mathbb{E} \left[ \frac{1}{m} \sum_{i=1}^m \omega_i^*A\omega_i \right] = \frac{1}{m} \sum_{i=1}^m \mathbb{E} \left[ \omega_i^* A \omega_i\right]. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-52eebd003a597cac5b2c69f7b6e537e8_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E} [\hat{\tr}] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-96f71a1b054ffed9e316cf9cb7a16c5c_l3.png "Rendered by QuickLaTeX.com") , it is sufficient to prove that
, it is sufficient to prove that ![\mathbb{E} \left[\omega_i^*A\omega_i\right] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-120169dca277175c51f09b6f320cf5fa_l3.png "Rendered by QuickLaTeX.com") for each
for each  matrix, then a number equals its trace,
matrix, then a number equals its trace,  . The second trick is the
. The second trick is the  matrix
matrix  and a
and a  matrix
matrix  , we have
, we have  . The cyclic property
. The cyclic property ![\[\tr[BCD] = \tr[(BC)D] = \tr[D(BC)] = \tr[DBC].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cc1cd4920a2431e93b5453b5373999e3_l3.png "Rendered by QuickLaTeX.com")
![\[\tr [BCD] \ne \tr[CBD] \quad \text{in general}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f563660fc58383a37ae7bedea16b2b9c_l3.png "Rendered by QuickLaTeX.com")
 as beads on a closed loop of string. One can move the last bead
as beads on a closed loop of string. One can move the last bead ![\tr [BCD] = \tr[DBC]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7656627b133add21ab8765532def07d6_l3.png "Rendered by QuickLaTeX.com") , but not interchange two beads,
, but not interchange two beads, ![\tr[BCD] \ne \tr[CBD]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0f43e4f72510a69b596f7b9f7504dd58_l3.png "Rendered by QuickLaTeX.com") .
.![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[A\omega_i\omega_i^*\right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b1bb373766e64179bc0635f058a07f3f_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \mathbb{E} \tr \left[A\omega_i\omega_i^*\right] = \tr(A \mathbb{E}[\omega_i\omega_i^*] ).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1d10f835c99efab005f0ad12cdb5647b_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} \left[\omega_i^*A\omega_i\right] = \tr(A \mathbb{E}[\omega_i\omega_i^*] ) = \tr(A\cdot I) = \tr A.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b51781498146df3547c1503e451ad682_l3.png "Rendered by QuickLaTeX.com")
 ‘s are isotropic (3) and now assume that
‘s are isotropic (3) and now assume that ![\[\Var(\hat{\tr}) = \frac{1}{m^2} \sum_{i=1}^m \Var(\omega_i^*A\omega_i).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d9398dc43178ba9bb72aead0214e77c8_l3.png "Rendered by QuickLaTeX.com")
 , we then get
, we then get![\[\Var(\hat{\tr}) = \frac{1}{m} \Var(\omega^*A\omega).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4501a18842234362f8f99d523c7c723e_l3.png "Rendered by QuickLaTeX.com")
 , which is characteristic of
, which is characteristic of  is unbiased (i.e, (3)), this means that the mean square error decays like
is unbiased (i.e, (3)), this means that the mean square error decays like ![\[\left| \hat{\tr} - \tr A \right| \lessapprox \frac{\mathrm{const}}{\sqrt{m}}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c735791b8fe88d627bc2ebad8137fb1e_l3.png "Rendered by QuickLaTeX.com")
 , I must do
, I must do  the work!
the work! and (root-mean-square) error at rates
and (root-mean-square) error at rates ![\[\Var(\hat{\tr}_{\text{H++ or X}}) \le \frac{\mathrm{const}}{m^2},\quad \left| \hat{\tr}_{\text{H++ or X}} - \tr A \right| \lessapprox \frac{\mathrm{const}}{m}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-87438d87094d3df389400b5f7abee24a_l3.png "Rendered by QuickLaTeX.com")
 that makes the variance of the single–sample estimate
that makes the variance of the single–sample estimate  as low as possible. In this section, we will provide several explicit formulas for the variance of the Girard–Hutchinson estimator. Derivations of these formulas will appear at the end of this post. These variance formulas help illuminate the benefits and drawbacks of different test vector distributions.
as low as possible. In this section, we will provide several explicit formulas for the variance of the Girard–Hutchinson estimator. Derivations of these formulas will appear at the end of this post. These variance formulas help illuminate the benefits and drawbacks of different test vector distributions. we use
we use  and
and  to denote the real and imaginary parts. The variance of a random complex number
to denote the real and imaginary parts. The variance of a random complex number  is
is![\[\Var(z) := \mathbb{E} |z - \mathbb{E} z|^2 = \Var(\Re z) + \Var(\Im z).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8f636667a7dbbb3217896a962de1d28c_l3.png "Rendered by QuickLaTeX.com")
![\[\left\|A\right\|_{\rm F}^2 = \sum_{i,j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e132e4b9029c41dbfb99cd0e588d8d70_l3.png "Rendered by QuickLaTeX.com")
 , we have
, we have![\[\left\|A\right\|_{\rm F}^2 = \sum_{i=1}^n \lambda_i^2. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06b272e602552353c020f6399347c748_l3.png "Rendered by QuickLaTeX.com")
 denotes the
denotes the  denotes the
denotes the  rather than the conjugate transpose
rather than the conjugate transpose  is a number, it is equal to its own transpose:
is a number, it is equal to its own transpose:![\[\omega^\top A \omega = (\omega^\top A \omega)^\top = \omega^\top A^\top \omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3d543dc0d4d64d3c28f0f291cd9c8e83_l3.png "Rendered by QuickLaTeX.com")
![\[\omega^\top A\omega = \frac{\omega^\top A \omega + \omega^\top A^\top \omega}{2} = \omega^\top \left( \frac{A + A^\top}{2} \right)\omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f1bbe4f0c983f73e2461b593b752f54a_l3.png "Rendered by QuickLaTeX.com")
 .
. .
.![\[\Var(\omega^\top A\omega) = 2 \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-091cf638fab943be5fd224acdb42668f_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\omega^\top A \omega) = 2\sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-74621dad79cb3f06a8b70c661df4e01d_l3.png "Rendered by QuickLaTeX.com")
 are uniformly distributed on the real sphere of radius
are uniformly distributed on the real sphere of radius  :
:  .
. ![\[\Var(\omega^\top A\omega) = \frac{2n}{n+2} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n} |\tr A|^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1e419b8da9b953a0b8b914df9e89f8c6_l3.png "Rendered by QuickLaTeX.com")
![\[A = A^{\rm H} + i A^{\rm SH}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d61da43cd6497ceefd2306aabb52ee67_l3.png "Rendered by QuickLaTeX.com")
![\[A^{\rm H} = \frac{A+A^*}{2} ,\quad A^{\rm SH} = \frac{A - A^*}{2i}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-dc287a49a59dca9421fdd2bef6be5334_l3.png "Rendered by QuickLaTeX.com")
 is skew-Hermitian). Since
is skew-Hermitian). Since  and
and  are both Hermitian, we have
are both Hermitian, we have![\[\Re(\omega^* A\omega) = \omega^* A^{\rm H} \omega, \quad \Im (\omega^* A \omega) = \omega^* A^{\rm SH} \omega.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6809a35cd06d92218ce8d597847f74bd_l3.png "Rendered by QuickLaTeX.com")
 can be broken into Hermitian and skew-Hermitian parts:
can be broken into Hermitian and skew-Hermitian parts:![\[\Var(\omega^* A\omega) = \Var(\omega^* A^{\rm H}\omega) + \Var(\omega^* A^{\rm SH}\omega).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e4658c38df479c5805dabd94b64cc0b3_l3.png "Rendered by QuickLaTeX.com")
 .
. for
for  standard normal random variables.
standard normal random variables. ![\[\Var(\omega^* A\omega) = \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1beb66e032d83431dac6f8e0d6655eec_l3.png "Rendered by QuickLaTeX.com")
 .
. ![\[\Var(\omega^* A \omega) = \sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1cb0e8df66c34056cd350752070af17a_l3.png "Rendered by QuickLaTeX.com")
 .
. ![\[\Var(\omega^* A\omega) = \frac{n}{n+1} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n} |\tr A|^2 \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-aacfa2afb45905f6abe0697c769cd1b7_l3.png "Rendered by QuickLaTeX.com")
 are isotropic (2), independent from each other, and have independent entries, then for any fixed real symmetric matrix
are isotropic (2), independent from each other, and have independent entries, then for any fixed real symmetric matrix  .
. -valued and an
-valued and an  -valued test vectors on unified footing, let
-valued test vectors on unified footing, let  denote either
denote either  ) or a complex Hermitian matrix (if
) or a complex Hermitian matrix (if  ). Let a
). Let a  of
of ![\[\text{If $A \in \mathscr{A}$ and $U$ is $\field$-unitary, then $U^*AU \in \mathscr{A}$.}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-18b4216d44b2364ea39ed4a078a067af_l3.png "Rendered by QuickLaTeX.com")
 is minimized by choosing
is minimized by choosing  .
. , the set of all
, the set of all  , where
, where  is a diagonal matrix reporting
is a diagonal matrix reporting  has the same distribution as
has the same distribution as  . Therefore,
. Therefore,![\[\Var(\omega^\top A \omega) = \Var(\omega^\top \Lambda \omega) = \Var \left( \sum_{i=1}^n \lambda_i \omega_i^2 \right) = \sum_{i=1}^n \lambda_i^2 \Var(\omega_i^2) = 2\sum_{i=1}^n \lambda_i^2 = 2\left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a7416823720bac922fae7cc685892a49_l3.png "Rendered by QuickLaTeX.com")
 . Thus,
. Thus,![\[\Var(\omega^\top A \omega) = \Var(\omega^\top \mathfrak{R}(A) \omega) + \Var(\omega^\top \mathfrak{I}(A) \omega) = 2\left\|\mathfrak{R}(A)\right\|_{\rm F}^2 + 2\left\|\mathfrak{I}(A)\right\|_{\rm F}^2 = 2\left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-527980eb3510f2c015106e6945126d41_l3.png "Rendered by QuickLaTeX.com")
![\[\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega] = \sum_{i,j=1}^n A_{ij} \omega_i\omega_j - \sum_{i=1}^n A_{ii} = \sum_{i\ne j} A_{ij} \omega_i\omega_j + \sum_{i=1}^n A_{ii}(\omega_i^2-1).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4489cc9cb97ee7a0896bc61193d79910_l3.png "Rendered by QuickLaTeX.com")
 for every
for every  can be replaced by two times the sum over
can be replaced by two times the sum over  :
:![\[\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega] = 2\sum_{i< j} A_{ij} \omega_i\omega_j.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-afc5f6bb40cd00b7897f25d73751f4ca_l3.png "Rendered by QuickLaTeX.com")
 are
are ![\[\Var(a_1 X_1+\cdots+a_kX_k) = |a_1|^2 \Var(X_1) + \cdots + |a_k|^2 \Var(X_k)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b6fd81d15f564f514f79ab4c33d133d8_l3.png "Rendered by QuickLaTeX.com")
 and numbers
and numbers  . Ergo,
. Ergo,![\begin{align*}\Var(\omega^\top A\omega) &= \Var(\omega^\top A \omega - \mathbb{E}[\omega^\top A \omega]) \\&= \Var\left(\sum_{i< j} 2A_{ij} \omega_i\omega_j\right) \\&= \sum_{i<j} 4 |A_{ij}|^2 \Var(\omega_i\omega_j) \\&= \sum_{i<j} 4 |A_{ij}|^2 \\&= 2 \sum_{i\ne j} |A_{ij}|^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bbb33f378fabd391c28d938420d3a32b_l3.png "Rendered by QuickLaTeX.com")
 is a uniform random sign, which has variance
is a uniform random sign, which has variance  camels, the second son took
camels, the second son took  camels, the third son
camels, the third son  camels and the wise man took his own camel and went away.
camels and the wise man took his own camel and went away. is easy. Fortunately, we already know such a distribution, the Gaussian distribution, for which we already calculated
is easy. Fortunately, we already know such a distribution, the Gaussian distribution, for which we already calculated  .
.![\[g = \sqrt{\frac{a}{n}} \omega,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-a93345cf94749f98e715e42bc796501c_l3.png "Rendered by QuickLaTeX.com")

 using the
using the ![\[\Var(g^\top A g) = \mathbb{E}[\Var(g^\top A g \mid a)] + \Var(\mathbb{E}[g^\top A g \mid a]).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e4e5ed235fbf5b2e026bcc58bd23becc_l3.png "Rendered by QuickLaTeX.com")
 and
and ![\mathbb{E}[ \cdot \mid a]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6035d77442675ac2f790cfb5417e1072_l3.png "Rendered by QuickLaTeX.com") denote the
denote the  and treating
and treating ![\begin{align*}\Var(g^\top A g) &= \mathbb{E}[\Var(a/n \cdot \omega^\top A \omega \mid a)] + \Var(\mathbb{E}[a/n \cdot \omega^\top A \omega \mid a]) \\&=\mathbb{E}[(a/n)^2\Var(\omega^\top A \omega)] + \Var(a/n \cdot \mathbb{E}[\omega^\top A \omega]) \\&= \frac{1}{n^2} \mathbb{E}[a^2] \cdot \Var(\omega^\top A \omega) + \frac{1}{n^2} \Var(a) |\mathbb{E} [\omega^\top A \omega]|^2.\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-be052caddf1d08941925fa0283784635_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[a^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-53042ba72cdd3991b14b5d4508da6564_l3.png "Rendered by QuickLaTeX.com") and
and  are known quantities that
are known quantities that ![\[\mathbb{E}[a^2] = n(n+2), \quad \Var(a) = 2n.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-59ecda0ea21d92e43e3182d5cc5a7217_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E} [\omega^\top A \omega] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-293908b9583df45d93349435d76b981a_l3.png "Rendered by QuickLaTeX.com") . Plugging these all in, we get
. Plugging these all in, we get![\[2\left\|A\right\|_{\rm F}^2 = \frac{n+2}{n} \Var(\omega^\top A\omega) + \frac{2}{n} |\tr A|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1c9b70fd4e5bc6ca4006114830d45619_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\omega^\top A\omega) = \frac{2n}{n+2} \left( \left\|A\right\|_{\rm F}^2 - \frac{1}{n}|\tr A|^2\right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7ab53e5895f9a1f474f5bf99fbec5662_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\omega^*A \omega) = \Var \left( \sum_{i=1}^n \lambda_i |\omega_i|^2 \right) = \sum_{i=1}^n \Var(|\omega_i|^2) \lambda_i^2 = \sum_{i=1}^n \lambda_i^2 = \left\|A\right\|_{\rm F}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-68f4685a4cd845b98fc1a8fd1494e00a_l3.png "Rendered by QuickLaTeX.com")
 is a
is a  ) reveals that
) reveals that![\[\Var\left( \omega^* A \omega \right) = \Var \left( \sum_{i<j} 2 \Re(A_{ij} \overline{\omega_i} \omega_j) \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b0e61723c22b056d1aca51694c407e6d_l3.png "Rendered by QuickLaTeX.com")
 are
are ![\[\Var\left( \omega^* A \omega \right) = \Var \left( \sum_{i<j} 2 \Re(A_{ij} \overline{\omega_i} \omega_j) \right) = 4\sum_{i<j} \Var \left( \Re(A_{ij} \overline{\omega_i} \omega_j) \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f2d77eb554a1e1ffbe4370efd8c704fb_l3.png "Rendered by QuickLaTeX.com")
 is uniformly distributed on the complex unit circle, we can assume without loss of generality that
is uniformly distributed on the complex unit circle, we can assume without loss of generality that  . Thus, letting
. Thus, letting  be uniform on the complex unit circle,
be uniform on the complex unit circle,![\[\Var\left( \omega^* A \omega \right) = 4\sum_{i<j} \Var \left( |A_{ij}|\Re(\phi)) \right) = 4\Var\left( \Re(\phi) \right)\sum_{i<j}|A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-76d2ce84714c4549a86686259bd96847_l3.png "Rendered by QuickLaTeX.com")
![\[1 = \Var(\phi) = \Var(\Re \phi) + \Var(\Im \phi) = 2 \Var(\Re \phi)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f83462ae4de80311482849de445edd5a_l3.png "Rendered by QuickLaTeX.com")
 . Thus
. Thus![\[\Var\left( \omega^* A \omega \right) = 2 \sum_{i<j}|A_{ij}|^2 = \sum_{i\ne j} |A_{ij}|^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-436eeb79ad0eddde0752f1a398397ad5_l3.png "Rendered by QuickLaTeX.com")
 and
and  denote the real and imaginary parts of a vector or matrix, taken entrywise. The key insight is that if
denote the real and imaginary parts of a vector or matrix, taken entrywise. The key insight is that if ![\[\mathscr{R}(\omega) := \twobyone{\mathfrak{R}(\omega)}{\mathfrak{I}(\omega)}\in\real^{2n} \quad \text{is a uniform random vector on the real sphere of radius $\sqrt{n}$}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0ff873eff96e863cf02d36e65a5ca1be_l3.png "Rendered by QuickLaTeX.com")
 .
. . To do this, recall that one way of representing complex numbers is by
. To do this, recall that one way of representing complex numbers is by  matrices:
matrices:![\[a + bi \iff \twobytwo{a}{-b}{b}{a}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-21b802836b33431568e5d854455d87d1_l3.png "Rendered by QuickLaTeX.com")
![\[\mathscr{R}(A) = \twobytwo{\mathfrak{R}(A)}{-\mathfrak{I}(A)}{\mathfrak{I}(A)}{\mathfrak{R}(A)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-12a37c4298c9e2d49dfa2f3ac32fe573_l3.png "Rendered by QuickLaTeX.com")
![\[\mathscr{R}(A + B) = \mathscr{R}(A) + \mathscr{R}(B), \quad \mathscr{R}(A\cdot B) = \mathscr{R}(A) \cdot \mathscr{R}(B)\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4e7fe7da4edb58f1ee1bb8a2b1d2a78b_l3.png "Rendered by QuickLaTeX.com")
 . A short calculation reveals
. A short calculation reveals![\[\omega^*A\omega = \mathscr{R}(\omega)^\top \mathscr{R}(A)\mathscr{R}(\omega) .\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d92927d567b8907621af83ab283672a5_l3.png "Rendered by QuickLaTeX.com")
 is a uniform random vector on the sphere of radius
is a uniform random vector on the sphere of radius  . Thus, by the variance formula for the real sphere, we get
. Thus, by the variance formula for the real sphere, we get![\[\Var(\omega^*A\omega) = \Var[(\sqrt{2}\mathscr{R}(\omega))^\top (\mathscr{R}(A)/2)(\sqrt{2}\mathscr{R}(\omega) )] = \frac{4n}{2n+2} \left[ \|\mathscr{R}(A)/2\|_{\rm F}^2 - \frac{1}{8n}(\tr\mathscr{R}(A))^2 \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ff16ba8dd015218ea642633d77f2a37a_l3.png "Rendered by QuickLaTeX.com")
 and
and  . Plugging this in, we obtain
. Plugging this in, we obtain![\[\Var(\omega^*A\omega)= \frac{n}{n+1} \left[ \|A\|_{\rm F}^2 - \frac{1}{n}(\tr A)^2 \right].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c9186eb813af3cd67173d73e32885a22_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E} [(\omega^*A \omega)^2].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-8e3e10402f389b06dcd42db97c5ed116_l3.png "Rendered by QuickLaTeX.com")
 . For those unfamiliar with the tensor product, the main properties we will be using
. For those unfamiliar with the tensor product, the main properties we will be using ![\[(A\otimes B) (C\otimes D) = (AB) \otimes (CD), \quad \tr(A\otimes B) = \tr A \cdot \tr B. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c5ea1a0235766ad15b67d02b47cfea2e_l3.png "Rendered by QuickLaTeX.com")
![\[\omega^* A \omega = \tr (\omega^*A\omega) = \tr (A \omega\omega^*).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-578bb521ecfb65fc4025f13431673b08_l3.png "Rendered by QuickLaTeX.com")
![\[(\omega^*A\omega)^2 = (\tr [A \omega\omega^*])^2 = \tr [A\omega\omega^* \otimes A\omega\omega^*] = \tr [(A\otimes A) (\omega\omega^* \otimes \omega\omega^*)].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-5161bc0d787be08130c4cfaed0c84489_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[(\omega^*A\omega)^2]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-98fe9a5e8220c22709c9d6ade6c3141b_l3.png "Rendered by QuickLaTeX.com") , it will be sufficient to evaluate
, it will be sufficient to evaluate ![\mathbb{E}[\omega\omega^* \otimes \omega\omega^*]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-06ac3d64041068176a86563cec4c67ba_l3.png "Rendered by QuickLaTeX.com") . Forunately, there is a useful formula for these expectation provided by a field of mathematics known as representation theory (see Lemma 1 in
. Forunately, there is a useful formula for these expectation provided by a field of mathematics known as representation theory (see Lemma 1 in ![\[\mathbb{E}[ \omega\omega^* \otimes \omega\omega^*] = \frac{2n}{n+1} \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e5eddca3275c6bbb82fa6b789efe7664_l3.png "Rendered by QuickLaTeX.com")
 is the
is the  . Therefore, we have that
. Therefore, we have that![\[\mathbb{E}[(\omega^*A\omega)^2] = \tr [(A\otimes A) \mathbb{E}(\omega\omega^* \otimes \omega\omega^*)] = \frac{2n}{n+1} \tr [(A\otimes A) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}].\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6c4f09e537808effd6c51c0aa4d20f6e_l3.png "Rendered by QuickLaTeX.com")
![\[\tr \left[(A\otimes B) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}\right] = \frac{1}{2} \left( \tr(AB) + \tr A \cdot \tr B \right).\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-3ea7660d7e72964cf11ef2239731a497_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\Var(\omega^* A \omega) &= \mathbb{E}[(\omega^*A\omega)^2] - (\mathbb{E}[\omega^*A\omega])^2 \\&= \frac{2n}{n+1}\tr [(A\otimes A) \operatorname{Proj}_{\operatorname{Sym}^2(\complex^n)}] - (\tr A)^2 \\&= \frac{n}{n+1}\left[ \tr A^2 + (\tr A)^2 \right] - (\tr A)^2 \\&= \frac{n}{n+1}\left[ \left\|A\right\|_{\rm F}^2 - \frac{1}{n} (\tr A)^2 \right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7af313e9e07f20b93b0de83867f20a04_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E}[\omega\omega^\top] = I\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-232c1e956304b14a7a6f4b55f9ba35e3_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[\omega_i\omega_j] = \delta_{ij}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-9986528214c0653217367aca2a90d568_l3.png "Rendered by QuickLaTeX.com") , where
, where  is the
is the ![\begin{align*}\mathbb{E}[ (\omega^*A \omega)^2] &= \mathbb{E}\left[ \left( \sum_{i=1}^n A_{ii} \omega_i^2 +2 \sum_{i<j} A_{ij}\omega_i\omega_j) \right)^2\right] \\&= \sum_{i=1}^n A_{ii}^2 \mathbb{E}[\omega_i^4] + \sum_{i<j} (2A_{ii}A_{jj}+4A_{ij}^2) \mathbb{E}[\omega_i^2]\mathbb{E}[\omega_j^2] \\&= \sum_{i=1}^n A_{ii}^2 \mathbb{E}[\omega_i^4] + \sum_{i<j} (2A_{ii}A_{jj}+4A_{ij}^2) .\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4115914a6e648438e66028437f38ef2a_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\omega^*A\omega) = \mathbb{E}[ (\omega^*A \omega)^2] - (\mathbb{E}[\omega^* A \omega])^2 = \sum_{i=1}^n A_{ii}^2 (\mathbb{E}[|\omega_i|^4]-1) + 4\sum_{i<j} A_{ij}^2.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-006e2eb8d5f2162654d8c2de39f5a8bc_l3.png "Rendered by QuickLaTeX.com")
 as small as possible. Since
as small as possible. Since  , the smallest possible value for
, the smallest possible value for  , which is obtained by populating
, which is obtained by populating ![\[\omega = a \cdot s \quad \text{for} \quad a \in [0,+\infty), \: s \in\{x\in \field^n : x^*x = n \}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-11867b7bff40d97d79a74d5136774fd9_l3.png "Rendered by QuickLaTeX.com")
![\[\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega}) \le \sup_{A\in\mathscr{A}} \Var(\omega^*A\omega \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e54c170954072838232ab5ff7c78cf25_l3.png "Rendered by QuickLaTeX.com")
![\[\tilde{\omega} = a\cdot t \quad \text{and}\quad t\sim \text{Uniform} \{ x \in \field^n :x^*x = n \} \quad \text{is independent of $a$}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-cb668b2c9f168bca81deb51a17726b1d_l3.png "Rendered by QuickLaTeX.com")
 for a uniformly random
for a uniformly random ![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[(\tilde{\omega}^*A\tilde{\omega})^2] - (\tr A)^2\right]\\&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-83564c7ba654655b42890c8adb65636f_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&= \sup_{A\in\mathscr{A}} \left[\mathbb{E}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right] \\&\le \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right].\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f182f68bcb3cbe40c8b44cf5b585e474_l3.png "Rendered by QuickLaTeX.com")
 for
for  is the same as the supremum of
is the same as the supremum of ![\begin{align*}\sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega})&\le \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*(Q^*AQ)s] - (\tr (Q^*AQ))^2\right] \\&= \mathbb{E}_Q \sup_{A\in\mathscr{A}} \left[\mathbb{E}_{a,s}[a^2 \cdot s^*As] - (\tr A)^2\right] \\&= \sup_{A\in\mathscr{A}} \Var(\omega^*A\omega).\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-bd5736cb133d33ed28ab0f2305ac9678_l3.png "Rendered by QuickLaTeX.com")
![\[\sup_{A\in\mathscr{A}} \Var(t^*At) \le \sup_{A\in\mathscr{A}} \Var(\tilde{\omega}^*A\tilde{\omega}), \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-0de32ccbfe09388b9d36757b7f140a8e_l3.png "Rendered by QuickLaTeX.com")
![\begin{align*}\Var(\tilde{\omega}^*A\tilde{\omega})&= \Var(a^2\cdot t^*At) \\&= \mathbb{E}[\Var(a^2\cdot t^* A t \mid a)] + \Var(\mathbb{E}[a^2\cdot t^* A t \mid a]) \\&= \mathbb{E}[a^4]\Var(t^* A t )+ (\tr A)^2\Var(a^2) \\&\ge \mathbb{E}[a^4]\Var(t^* A t ).\end{align*}](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c74465c627f5e70f943ce415fbfafd22_l3.png "Rendered by QuickLaTeX.com")
![\mathbb{E}[t^*At] = \tr A](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6ee9ad07ec8316cc6003116e3c311189_l3.png "Rendered by QuickLaTeX.com") . By definition,
. By definition, ![\[\mathbb{E}[a^2] = \frac{1}{n}\mathbb{E}[\omega^*\omega] = \frac{1}{n} \mathbb{E}[\tr (\omega\omega^*)] = \frac{1}{n} \tr (\mathbb{E}[\omega\omega^*]) = \frac{\tr I}{n} = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4da8a9a33ffb6559da8625435549f7c2_l3.png "Rendered by QuickLaTeX.com")
![\[\mathbb{E}[a^4] = \mathbb{E}[(a^2)^2] \ge (\mathbb{E}[a^2])^2 = 1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-356fd56d5d04dbda45fa992ae083be0f_l3.png "Rendered by QuickLaTeX.com")
![\[\Var(\tilde{\omega}^*A\tilde{\omega}) \ge \mathbb{E}[a^4]\Var(t^* A t ) \ge \Var(t^*At) \quad \text{for every }A,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-fc9331e3508e9102014ecc83f4517cc5_l3.png "Rendered by QuickLaTeX.com")
 -decompositions, sparse component analysis, and the blind separation of sums of exponentials
-decompositions, sparse component analysis, and the blind separation of sums of exponentials![\[f(t) = a_0 \cos(\omega_0 t - \phi_0) + a_1 \cos(\omega_1 t - \phi_1) + a_2 \cos(\omega_2 t - \phi_2). \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-20caba029479063b3c7d59617eff2225_l3.png "Rendered by QuickLaTeX.com")
 ,
,  , and
, and  .
. . In practice, we only have access to the function
. In practice, we only have access to the function  which we assume are equally spaced
which we assume are equally spaced![\[t_j = j\Delta t \quad \textnormal{for} \quad j = 0,1,2,\ldots,n-1.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4ad3ecc9f18da4793ab2a0113f4afae8_l3.png "Rendered by QuickLaTeX.com")
![\[f_j = f(t_j) \quad \textnormal{for} \quad j = 0,1,2,\ldots,n-1\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-2945d5d9092cf48b8b7503194ef8d8e0_l3.png "Rendered by QuickLaTeX.com")
 , where
, where  it will be helpful to rewrite our formula (1) for
it will be helpful to rewrite our formula (1) for ![\[\cos \alpha = \frac{\mathrm{e}^{\mathrm{i} \alpha}+\mathrm{e}^{-\mathrm{i} \alpha}}{2}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-1004ce391e777282f5b1cac7ea69b7b6_l3.png "Rendered by QuickLaTeX.com")
![\[a_0 \cos(\omega_0 t - \phi_0) = d_0 \mathrm{e}^{\mathrm{i} \omega_0t} + d_1 \mathrm{e}^{-\mathrm{i} \omega_0t}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b477beb50968f3d0ef261aeafaef89f1_l3.png "Rendered by QuickLaTeX.com")
 and
and  are complex coefficients
are complex coefficients  and
and  which contain the same information as the original parameters
which contain the same information as the original parameters  and
and  . Now notice that we are only interest in values
. Now notice that we are only interest in values  which are multiples of the spacing
which are multiples of the spacing  . Thus, our frequency component can be further rewritten as
. Thus, our frequency component can be further rewritten as![\[a_0 \cos(\omega_0 t_j - \phi_0) = d_0 z_0^j + d_1 z_1^j\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f3728deaec66b4719f2d79377295a1a0_l3.png "Rendered by QuickLaTeX.com")
 and
and  . Performing these reductions, our samples
. Performing these reductions, our samples  take the form
take the form![\[f_j = d_0 z_0^j + d_1 z_1^j + \cdots + d_5 z_5^j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-80d74d6613b6c1dd28e8e39741fe2043_l3.png "Rendered by QuickLaTeX.com")
 and
and  in the relation (2) from a small number of measurements
in the relation (2) from a small number of measurements  . This can reveal patterns in the sequence which are less obvious when it is represented as given just as numbers. For instance, any seven columns of
. This can reveal patterns in the sequence which are less obvious when it is represented as given just as numbers. For instance, any seven columns of  which can be much larger than seven. In addition, as we will soon effectively exploit later, vectors in the nullspace of
which can be much larger than seven. In addition, as we will soon effectively exploit later, vectors in the nullspace of ![\[H = \begin{bmatrix} f_0 & f_1 & f_2 & \cdots & f_{(n-1)/2} \\f_1 & f_2 & f_3 & \cdots & f_{(n-1)/2+1} \\f_2 & f_3 & f_4 & \cdots & f_{(n-1)/2+2} \\\vdots & \vdots & \vdots & \ddots & \vdots \\f_{(n-1)/2} & f{(n-1)/2+1} & f{(n-1)/2+2} & \cdots & f_{n-1} \end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-e58f400c13a096efb5c1682c52003056_l3.png "Rendered by QuickLaTeX.com")
 . Observe that the entry in position
. Observe that the entry in position  . (We use a
. (We use a  .)
.) using (2):
using (2):![\[H_{ij} = f_{i+j} = \sum_{k=0}^5 d_k z_k^{i+j} = \sum_{k=0}^5 d_k z_k^i \cdot z_k^j. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-c66cda6c8df0e635353009a6469c9d13_l3.png "Rendered by QuickLaTeX.com")
 was just begging to be factorized as
was just begging to be factorized as  , which we did. Equation (4) almost looks like the formula for the product of two matrices with entries
, which we did. Equation (4) almost looks like the formula for the product of two matrices with entries  , so it makes sense to introduce the
, so it makes sense to introduce the  matrix
matrix  . This is a so-called
. This is a so-called ![\[V = \begin{bmatrix}z_0^0 & z_0^1 & z_0^2 & \cdots & z_0^{(n-1)/2} \\z_1^0 & z_1^1 & z_1^2 & \cdots & z_1^{(n-1)/2} \\\vdots & \vdots & \vdots & \ddots & \vdots \\z_5^0 & z_5^1 & z_5^2 & \cdots & z_5^{(n-1)/2}\end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-52248d150045d64416cf49a569abf156_l3.png "Rendered by QuickLaTeX.com")
 diagonal matrix
diagonal matrix  , the formula (4) for
, the formula (4) for  , the factor
, the factor ![\[H = V^\top D V. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-37a707b3000f4e0e2be34695da3f0f68_l3.png "Rendered by QuickLaTeX.com")
 describing our sampled recording
describing our sampled recording  , determining all the frequencies would be very straightforward.
, determining all the frequencies would be very straightforward. of
of ![\[f_m = c_{k-1} f_{m-1} + \cdots + c_1f_{m-k+1}+ c_0 f_{m-k} \quad \textnormal{for every} \quad m = k,\,{k+1},\,{k+2},\cdots. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-f01bd866e8c2df02dc870f3a78958c17_l3.png "Rendered by QuickLaTeX.com")
 because there are six terms in the formula (2) for
because there are six terms in the formula (2) for  , such a recurrence will allow us to determine the parameters
, such a recurrence will allow us to determine the parameters  . A very good rule of thumb in applied mathematics is to always write down linear equations in matrix–vector notation in see how it looks. Doing this, we obtain
. A very good rule of thumb in applied mathematics is to always write down linear equations in matrix–vector notation in see how it looks. Doing this, we obtain![\[\begin{bmatrix} f_6 \\ f_7 \\ \vdots \\ f_{n-1} \end{bmatrix} = \underbrace{\begin{bmatrix} f_0 & f_1 & f_2 & f_3 & f_4 & f_5 \\ f_1 & f_2 & f_3 & f_4 & f_5 & f_6 \\ \vdots & \vdots & \vdots & \vdots & \vdots & \vdots \\ f_{n-7} & f_{n-6} & f_{n-5} & f_{n-4} & f_{n-3} & f_{n-2} \end{bmatrix}}_{=F}\begin{bmatrix} c_0 \\ c_1 \\ \vdots \\ c_5 \end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-7d7dc3e345c47a62fa233007d8d0db69_l3.png "Rendered by QuickLaTeX.com")
 ,
,  going forward.
going forward. would also be fine for our purposes, but we assume
would also be fine for our purposes, but we assume ![\[f_{6 \, :\, n-1} = F c, \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8b53d6de5c09e51040ae688d9bc7d67_l3.png "Rendered by QuickLaTeX.com")
 for the vector on the left-hand side of (7) and collected the recurrence coefficients
for the vector on the left-hand side of (7) and collected the recurrence coefficients  . For a typical system of linear equations like (8), we would predict the system to have no solution
. For a typical system of linear equations like (8), we would predict the system to have no solution  ), the system equations (8) has more equations than unknowns. Fortunately, we are not in the typical case. Despite the fact that we have more equations than unknowns, the linear equations (8) have a unique solution
), the system equations (8) has more equations than unknowns. Fortunately, we are not in the typical case. Despite the fact that we have more equations than unknowns, the linear equations (8) have a unique solution  In particular, the matrix on the right-hand side of this equation is guaranteed to be nonsingular under our assumptions. Using the Vandermonde decomposition, can you see why?
In particular, the matrix on the right-hand side of this equation is guaranteed to be nonsingular under our assumptions. Using the Vandermonde decomposition, can you see why? ; we seek to find all complex numbers
; we seek to find all complex numbers  gives
gives![\[z^6 = c_0 z^5 + c_1 z^4 + \cdots + c_6. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4fb9f7123e927dbb902df1eef680ef98_l3.png "Rendered by QuickLaTeX.com")
![\[\begin{bmatrix}f_0 \\ f_1 \\ \vdots \\ f_{n-1}\end{bmatrix} = \begin{bmatrix}1 & 1 & \cdots & 1 \\ z_0 & z_1 & \cdots & z_5 \\ \vdots & \vdots & \ddots & \vdots \\ z_0^{n-1} & z_1^{n-1} & \cdots & z_5^{n-1}\end{bmatrix} \begin{bmatrix}d_0 \\ d_1 \\ \vdots \\ d_{n-1}.\end{bmatrix}. \]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-4b18fe5a64350989e4534f4b08ef4c23_l3.png "Rendered by QuickLaTeX.com")
 . So far, the benefits of the matrix factorization perspective have yet to really reveal themselves.
. So far, the benefits of the matrix factorization perspective have yet to really reveal themselves. needed to describe the samples and the representation of the samples as a mixture of exponentials is uniquely determined only if the matrix
needed to describe the samples and the representation of the samples as a mixture of exponentials is uniquely determined only if the matrix  ,
,  , and
, and  . (Here, the superscript denotes an index rather than differentiation.) Each signal is a weighted mixture of three sources
. (Here, the superscript denotes an index rather than differentiation.) Each signal is a weighted mixture of three sources  ,
,  , and
, and  , each of which plays a musical chord of three notes (thus representable as a sum of cosines as in (1)). One can think of the sources of being produced three different musical instruments at different places in a room and the recordings
, each of which plays a musical chord of three notes (thus representable as a sum of cosines as in (1)). One can think of the sources of being produced three different musical instruments at different places in a room and the recordings  for each recording
for each recording  and form each collection of samples into a Hankel matrix
and form each collection of samples into a Hankel matrix![\[H^{(\ell)} = \begin{bmatrix} f_0^{(\ell)} & f_1^{(\ell)} & f_2^{(\ell)} & \cdots & f_{(n-1)/2}^{(\ell)} \\f_1^{(\ell)} & f_2^{(\ell)} & f_3^{(\ell)} & \cdots & f_{(n-1)/2+1}^{(\ell)} \\f_2^{(\ell)} & f_3^{(\ell)} & f_4^{(\ell)} & \cdots & f_{(n-1)/2+2}^{(\ell)} \\\vdots & \vdots & \vdots & \ddots & \vdots \\f_{(n-1)/2}^{(\ell)} & f_{(n-1)/2+1}^{(\ell)} & f_{(n-1)/2+2}^{(\ell)} & \cdots & f_{n-1}^{(\ell)} \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-426cddcdbfa1b1bae2c3aaccd49f9974_l3.png "Rendered by QuickLaTeX.com")
 ,
,  , and
, and  as slices of a
as slices of a  array given by
array given by![\[\mathcal{H}(:,:,\ell) = H^{(\ell)} \quad \textnormal{for} \quad \ell=1,2,3.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-b8b73fa4217e9c667877693a2f3e8348_l3.png "Rendered by QuickLaTeX.com")
![\[H = V^\top DV = \tilde{V}^\top \tilde{D} \tilde{V}\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-961c06b840f52d60a6fbc789026074a1_l3.png "Rendered by QuickLaTeX.com")
 can be uniquely determined from the measurements
can be uniquely determined from the measurements  rows,
rows,  and perhaps
and perhaps  ,
,  , etc.
, etc. This matrix has rank-two but no rank-two confluent Vandermonde decomposition. The issue is that when extended to an infinite Hankel matrix
This matrix has rank-two but no rank-two confluent Vandermonde decomposition. The issue is that when extended to an infinite Hankel matrix  this (infinite!) matrix has a rank exceeding the size of the original Hankel matrix
this (infinite!) matrix has a rank exceeding the size of the original Hankel matrix  into a Hankel matrix, it can also be useful to form them into a
into a Hankel matrix, it can also be useful to form them into a ![\[T = \begin{bmatrix} f_{(n-1)/2} & f_{(n-1)/2+1} & f_{(n-1)/2+2} & \cdots & f_{n-1} \\\\f_{(n-1)/2-1} & f_{(n-1)/2} & f_{(n-1)/2+1} & \cdots & f_{n-2} \\\\f_{(n-1)/2-2} & f_{(n-1)/2-1} & f_{(n-1)/2} & \cdots & f_{n-3} \\\\\vdots & \vdots & \vdots & \ddots & \vdots \\\\ f_0 & f_1 & f_2 & \cdots & f_{(n-1)/2} \end{bmatrix}.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-d576130eb33a8d98958a967ae78ae988_l3.png "Rendered by QuickLaTeX.com")
 has the appealing propery that the matrix–vector product
has the appealing propery that the matrix–vector product  computes a
computes a ![\[T = \operatorname{ReversedRows}(V^\top) \cdot DV.\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-6cde21a8060919d23383e6cb5fcbaebe_l3.png "Rendered by QuickLaTeX.com")
![\[T = V^* D V,\]](https://www.ethanepperly.com/wp-content/ql-cache/quicklatex.com-ef01b1f93fbb19fa844a5e40b1c8ceb7_l3.png "Rendered by QuickLaTeX.com")
 which are
which are  where
where  .
.
 real
real , one can define its
, one can define its  , to be the
, to be the  , one can extract a
, one can extract a  . If we instead consider the row space
. If we instead consider the row space  of
of 
 , as is exemplified when
, as is exemplified when  , we must also account for rows of
, we must also account for rows of  which cannot be written as linear combinations of the rows above, no matter how
which cannot be written as linear combinations of the rows above, no matter how  such rows, leading to the bound
such rows, leading to the bound
 the minimal rank for this completion problem.
the minimal rank for this completion problem. and
and  of the matrices
of the matrices  is again a vector subspace, and we choose a basis
is again a vector subspace, and we choose a basis  for it. The subspace
for it. The subspace  to it so that the enlarged matrix
to it so that the enlarged matrix  forms a basis for
forms a basis for  to add to
to add to  forms a basis for
forms a basis for  forms a basis for
forms a basis for  such that
such that  —in fact, this exact
—in fact, this exact  . Since
. Since  . Thus, doing the same with
. Thus, doing the same with 
 for the intersection of
for the intersection of  . We then extend these to bases
. We then extend these to bases  and
and  for
for  and
and  . From here, we derive the existence of matrix factorizations analogous to Eq. (4):
. From here, we derive the existence of matrix factorizations analogous to Eq. (4):
 . Applying this fact to the bases
. Applying this fact to the bases  and
and  for
for  such that
such that
 achieves the minimum possible rank
achieves the minimum possible rank  using the building blocks we’ve already built
using the building blocks we’ve already built
 entries whose value we have yet to determine. For us to have a hope of representing the matrix
entries whose value we have yet to determine. For us to have a hope of representing the matrix  into our rank factorization. Doing exactly this, we get the partially specified rank factorization
into our rank factorization. Doing exactly this, we get the partially specified rank factorization
 . From Eqs. (5) and (6), we deduce that
. From Eqs. (5) and (6), we deduce that  . Thus, we can fill in more entries:
. Thus, we can fill in more entries:
 and
and  to these free variables, we conclude that
to these free variables, we conclude that 
